


为了使战斗机尾部喷管、导弹弹头等飞行器的高温部位躲避雷达探测[1~3],必须使用具有高温电磁损耗性能和力学结构稳定的吸波材料。碳化硅(SiC)耐高温、耐腐蚀、高强度和抗氧化性能良好[4~6],是极具应用潜力的高温吸波材料。对SiC纳米颗粒掺杂N[7]、改变SiC纳米线的堆垛层错状态[8]或在SiC中复合磁性材料[9],都能提高其吸波性能。用化学气相沉积法(CVD)[10]、箔压法(FFF)[11~13]、PIP法[7,14]、碳热还原法[15,16]、冷冻干燥法[7,17]以及3D打印法[18~21]等工艺制备的SiC材料,其吸波性能良好。但是,SiC吸波粉剂和SiC纤维的制造成本高昂。鉴于此,本文用一种低成本原位反应法制备SiC吸波材料,研究原位反应温度对其物相、微观结构和吸波性能的影响及其机理。
1 实验方法
1.1 SiC吸波材料的制备
先将28.8 g酚醛树脂(-[-CH2-C6H4O-] n -,99%浓度)和1.7 g对甲苯磺酸(CH3C6H4SO3H,分析纯)溶解在无水乙醇(CH3CH2OH,分析纯)中,添加硅粉(1500目,分析纯) 25 g后以100 r/min的速度球磨30 min使其充分弥散混合,然后以3℃/min速度加热到60℃,烘至凝固后放入模具中,在40 kPa的压力下加热到90℃保温2 h,然后在120℃固化30 min。将固化物置于管式热解炉中在氮气保护下以5℃/min速度加热至700℃,保温1 h后自然冷却到室温。
将上述热解后的三种试样分别在温度为1800℃、2000℃和2200℃的高温真空炉中反应烧结1 h,冷却至室温后取出置于马弗炉中,以8℃/min速度加热至850℃保温30 min以除去残余碳,得到三种碳化硅试样,分别记为T1800、T2000和T2200。原位反应法制备SiC材料的流程图,在图1中给出。
图1

图1
原位反应法制备碳化硅材料的流程
Fig.1
Flow chart of in-situ reaction method for preparing silicon carbide materials
1.2 SiC吸波材料的表征
用扫描电子显微镜(SEM,SU-70,Hitachi,Japan)观察T1800、T2000、T2200试样的表面微观形貌;将试样研磨成细粉,用X射线衍射仪(XRD,D/Max2500PC,Rigaku,Japan)分析试样的物相,扫描速度为5°/min;用透射电子显微镜(TEM,Tecnai20,FEI,America)再进一步观察试样的微观形貌并分析晶体结构。
用矢量网络分析仪(VNA,N5230A,Agilent,America)测量SiC试样在X波段(8.2 GHz~12.4 GHz)的复介电常数(ε′,ε″)。
2 结果和讨论
2.1 碳化硅试样的形貌
图2给出了三种碳化硅试样的SEM照片。一方面,树脂在热解时分解生成大量小分子产生质量损失;另一方面,加入的硅粉渗入碳中参与原位反应起到了造孔作用。这些反应,使生成的SiC中出现大量的细微孔洞(图2a)。T1800试样的微孔较多,并在反应烧结温度1800℃生成了较小的SiC颗粒;由图2b可见,T2000的SiC颗粒比T1800的SiC颗粒明显的大,且颗粒之间互相联结;T2200的SiC颗粒继续变大(图2c),颗粒之间继续联结,形成较大的板状形貌。图2d给出了T2000 SiC晶粒表面形态,可见晶粒表面有许多级联结构。根据晶粒的生长条件,晶粒的生长机制应该是气相输运,即蒸发—冷凝过程。图2d给出的级联结构正是蒸发—冷凝过程的典型现象[22,23]。在2000℃反应烧结时发生了气相输运过程并随着反应烧结温度的升高生成的SiC颗粒联结变大。其原因是,SiC生成、气相输运和相变发生了如下的反应过程[23,24]
图2

图2
SiC试样的SEM表征
Fig.2
SEM characterization of SiC samples (a) SEM image of the T1800 specimen, (b) SEM image of the T2000 specimen, (c) SEM image of the T2200 specimen, (d) typical surface morphology of SiC grain growth at 2000oC
在温度高于1600℃时,硅粉与碳迅速反应生成立方晶型结构的β相碳化硅,即β-SiC。β-SiC在室温下是亚稳相,SiC的一种低温稳定形态。随着反应烧结温度的升高,一方面,少量的β-SiC与烧结炉中的微弱氧作用反应生成SiO2;另一方面,反应烧结温度不低于于2000℃时,β-SiC变得不稳定而易分解成SiC2和Si2C亚气相。SiO2与不稳定的β-SiC接触反应生成SiO蒸气,而不稳定的β-SiC提供碳的悬挂键与生成的SiO气体发生反应生成α-SiC晶核。β-SiC在高温下不稳定,β-SiC的总蒸汽压高于α-SiC的总蒸汽压。根据蒸汽压差,从β-SiC中蒸发的Si2C和SiC2不断扩散到α-SiC晶核并凝结,使烧结初期生成的α-SiC晶核迅速生长,消耗了不稳定的β-SiC。在这个过程的最后,β-SiC的过渡气态不断在α-SiC上凝结生成相互连接的较大多孔的板状结构[22,23,25~33]。
2.2 T1800、T2000和T2200试样的相组成
图3给出了三种试样的XRD谱。由图3可见,T1800试样在2θ角35.7°、41.4°、59.8°和71.8°处只出现β-SiC的衍射峰,表明在1800℃反应烧结生成的碳化硅仅由β-SiC构成。β-SiC所有的衍射峰都与α-SiC的衍射峰重合,但是图3中T2000试样在2θ角33.9°和37.9°处出现了只属于α-SiC的衍射峰。这表明,在2000℃,β-SiC开始相变反应生成α-SiC。T2200试样在2θ角34.2°和38.1°处的α-SiC衍射峰,比T2000试样的则更加突出。确定物相后查出对应的I值,并根据标准卡片(PDF74-2307)和(PDF75-1541)查得β-SiC、α-SiC的RIR值(K值),带入
图3
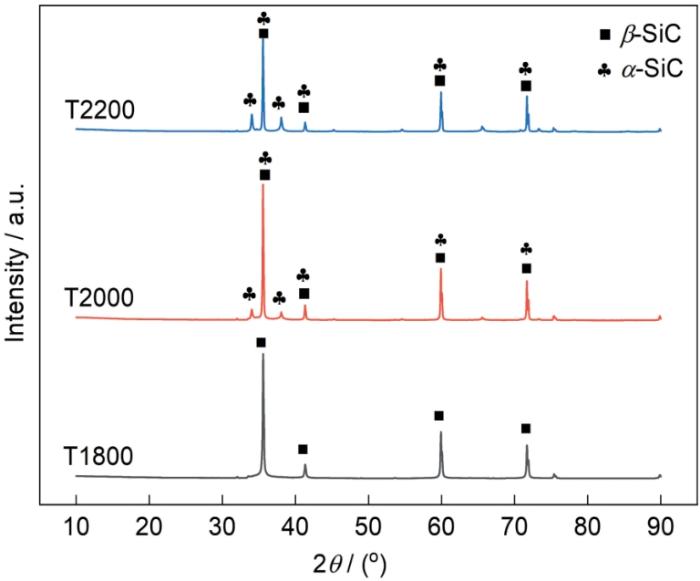
图3
不同反应烧结温度的SiC试样的XRD谱
Fig.3
XRD patterns of SiC samples with different reaction sintering temperatures
表1 T2000#和T2200#样品的定量分析结果
Table 1
| I(hight) | RIR(K) | Wi | |
|---|---|---|---|
| T2000#β-SiC | 385180 | 3.53 | 79.7% |
| T2000#α-SiC | 36995 | 1.33 | 20.3% |
| T2200#β-SiC | 248248 | 3.53 | 56.6% |
| T2200#α-SiC | 71587 | 1.33 | 43.4% |
表1中T2000试样中β-SiC的占比为79.7%,α-SiC的占比为20.3%,T2200试样中β-SiC的占比为56.6%,α-SiC的占比为43.4%。温度升高后,β-SiC的含量降低,α-SiC的含量提高。这表明,在2200℃反应烧结温度SiC试样走的β-SiC转变为α-SiC的程度比在2000℃时进一步加剧。
XRD测试结果表明,在原位反应烧结温度达到和高于2000℃时确实发生了SiC相变反应。
2.3 试样内部的微观晶体结构
图4给出了T1800试样的TEM表征结果,其中图4a是低倍照片,图4b是高倍照片,图4c是图4a中的选区电子衍射图,图4d是图4b中的选区放大图。对图4c的分析结果表明,该选区晶体衍射的三个晶面(111)、(220)和(
图4

图4
T1800试样的TEM表征
Fig.4
TEM characterization of T1800 sample (a) low-magnification image of the T1800 specimen, (b) high-magnification image of the T1800 specimen, (c) selected area electron diffraction pattern of figure a, (d) enlarged view of selected area of figure b
图5给出了T2000试样的TEM表征结果,其中图5a为低倍照片,图5b为高倍照片,右上角为图5b的傅里叶变换(FFT),图5c为图5a的选区电子衍射图,图5d为图5b的选区放大。分析图5b中的FFT图片的主衍射点晶面间距[35],测得其值为0.26 nm,与(PDF00-022-1273)标准卡片对比表明,T2000试样对应6H-SiC的(101)晶面;分析图5c可知,该选区晶体衍射的三个晶面(111)、(200)和(
图5

图5
T2000试样的TEM表征
Fig.5
TEM characterization of T2000 sample: (a) low-magnification image of the T2000 specimen, (b) high power diagram of T2000 sample, the upper right corner is the FFT diagram of b diagram, (c) selected area electron diffraction pattern of figure a, (d) enlarged view of the selected area of figure b
图6给出了T2200试样的TEM表征结果,其中图6a为T2200的低倍照片,图6b为T2200试样的高倍照片,其右上角为图6b的FFT图,图6c为图6a的选区电子衍射图,图6d为图6b的选区放大图。分析图6b中FFT图片的主衍射点晶面间距[35],测得其值为0.26 nm,与(PDF00-022-1273)标准卡片对比表明,T2200试样也对应6H-SiC的(101)晶面;分析图6c的结果表明,该选区晶体衍射出的三个晶面(104)、(110)和(
图6

图6
T2200试样的TEM表征
Fig.6
TEM characterization of T2200 sample: (a) low-magnification image of the T2200 specimen, (b) high power diagram of T2200 sample, the upper right corner is the FFT diagram of b diagram, (c) selected area electron diffraction pattern of figure a, (d) enlarged view of the selected area of figure b
综上所述,在用原位反应法制备SiC材料的过程中,反应烧结温度为1800℃时硅粉和热解碳反应生成3C结构的β相SiC;反应烧结温度为2000℃时,在蒸发—冷凝过程中3C结构的β相SiC向6H结构的α相SiC转变;在反应烧结温度2200℃该相变进一步加剧。这表明,控制反应烧结温度可调控SiC材料中β-SiC和α-SiC的比例,从而影响SiC材料的吸波性能。
2.4 SiC吸波材料的介电性能
图7给出了了T1800、T2000、T2200试样的复介电常数ε′、ε″和介电损耗角正切值。介电常数实部ε′表征材料储存电磁波电场的能力,介电常数虚部ε″与材料的电磁损耗相关[32]。结果表明,在1800℃制备的样品复介电常数最高,在2000℃制备的样品复介电常数次之、在2200℃制备的样品复介电常数最低。这表明,反应烧结温度越高制备的样品介电常数ε′和ε″越低。其原因是,SiC中的载流子在电磁场作用下运动产生极化,与晶格摩擦产生热能消耗了电磁波的能量,而β-SiC晶格结构不如α-SiC晶格结构稳定,晶格缺陷较多(空穴及掺杂),载流子浓度高于α-SiC,所以β-SiC对SiC的复介电常数产生重要影响。在较高的温度烧结时,SiC中的β-SiC向α-SiC转化,α-SiC的占比提高(图7a、b),SiC的实部和虚部都减小,并且介电常数的虚部比实部下降更多。这表明,反应烧结温度影响相变程度进而影响SiC的介电性能。
图7
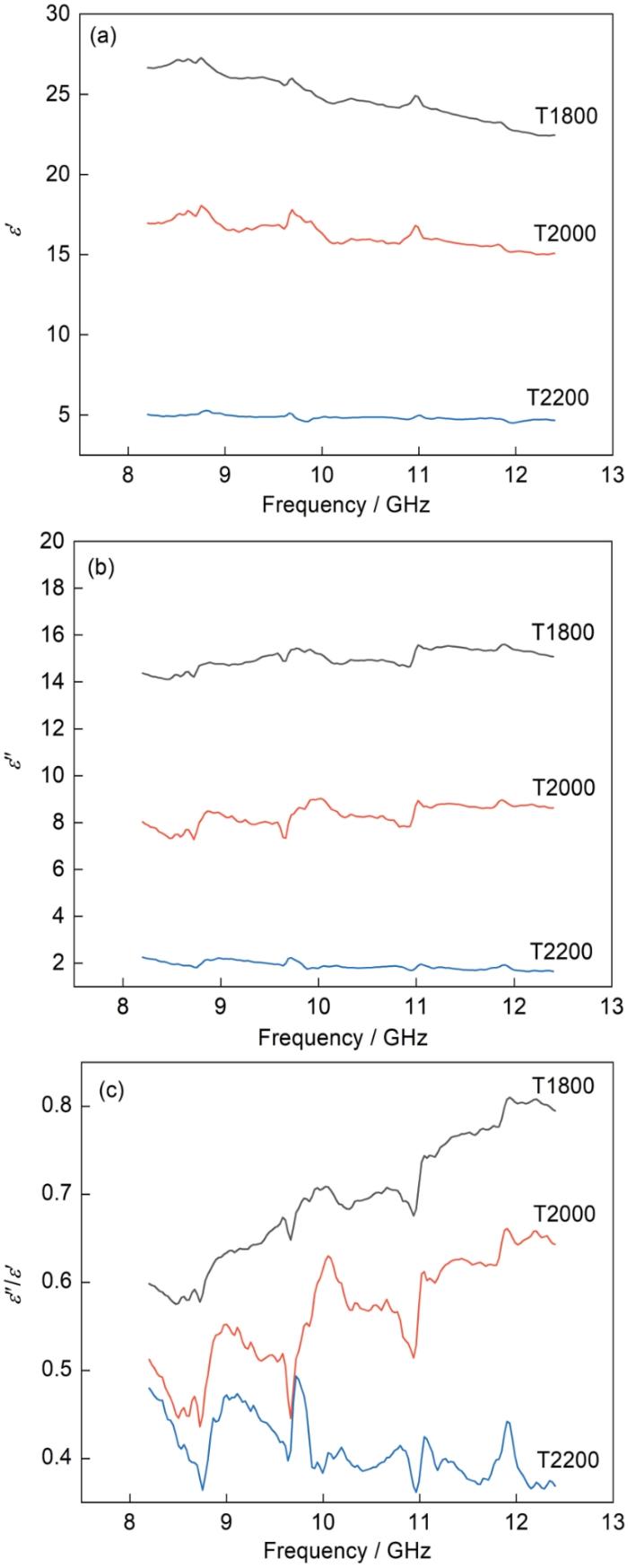
图7
SiC吸波材料的介电常数
Fig.7
Dielectric constant of SiC absorbing material (a) real part of dielectric constant, (b) imaginary part of dielectric constant, (c) loss angle tangent ε″/ε′
ε″/ε′可量化表征吸波材料的介电损耗,介电损耗角的正切值越大表明其介电损耗性能越好。根据ε′和ε″的实验值可计算tanδ = ε″/ε′,其中ε′为介电常数实部,表征材料对电磁波电场的储存能力;ε″为介电常数虚部,与材料的电磁损耗相关;δ称为介电损耗角[32]。如图7c所示,T1800、T2000和T2200试样的tanδ随着反应温度的提高呈明显降低的趋势,与复介电常数的变化规律相同。随着频率的提高T1800和T2000试样的tanδ有明显升高的趋势,而T2200试样的tanδ的变化趋势却并不明显。其原因是,SiC材料内部的载流子和官能团对低频和高频的响应程度不同。但是T2200试样的介电损耗曲线并没有出现这种情况,其原因可能是大量的β-SiC转化成α-SiC,使C和Si原子的堆垛方式更严密,晶格结构变得更稳定。
β-SiC的载流子比α-SiC的多,因此β-SiC对电磁波的介电损耗性能更高。但是,介电损耗能力过强导致材料的阻抗匹配性能变差,电磁波无法进入吸波材料内部。这表明,SiC与电磁波的阻抗必须匹配。SiC由β相转变成为α相使载流子浓度降低,介电损耗性能和阻抗匹配性能也随之发生变化。
2.5 SiC吸波材料的阻抗匹配
吸波材料的本征阻抗系数Z反映电磁波的进入程度。SiC的本征阻抗系数为
式中Z为本征阻抗系数,εr和µr为吸波材料的复介电常数和复磁导率。根据匹配理论,Z越趋近1则电磁波在材料表面的反射越少,表明材料的阻抗匹配性能越好[37]。根据图8给出的三个试样的阻抗匹配系数Z与频率的关系,T1800试样的本征阻抗系数为0.1~0.2,T2000试样的本征阻抗系数为0.23~0.24,T2200试样的本征阻抗系数为0.43~0.45。T1800试样的本征阻抗系数与1相差最大,表示入射到T1800材料表面的电磁波反射较多,不容易进入材料内部,即对微波的吸收性能较差;与T1800试样相比,电磁波更容易进入T2000试样内部。根据阻抗匹配,电磁波最容易进入T2200试样内部。这表明,烧结温度越高制备的SiC其阻抗系数越趋近1,更多的微波能进入其内。
图8
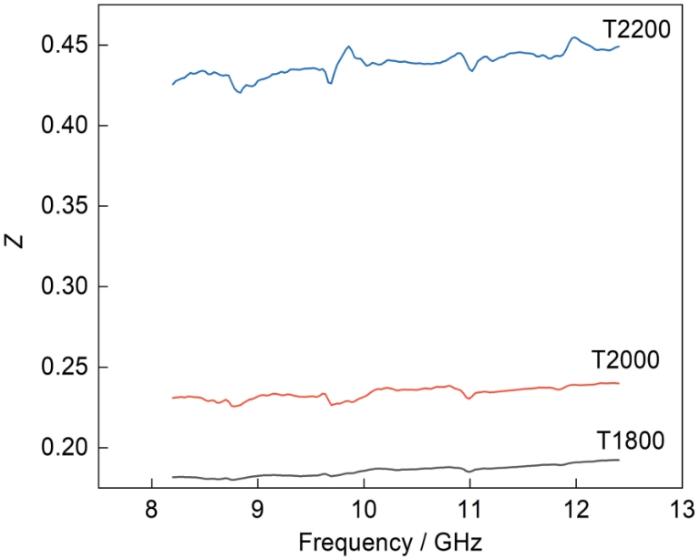
图8
试样T1800、T2000、T2200的本征阻抗
Fig.8
Intrinsic impedance of samples T1800, T2000,T2200
2.6 SiC吸波材料的反射率
图9
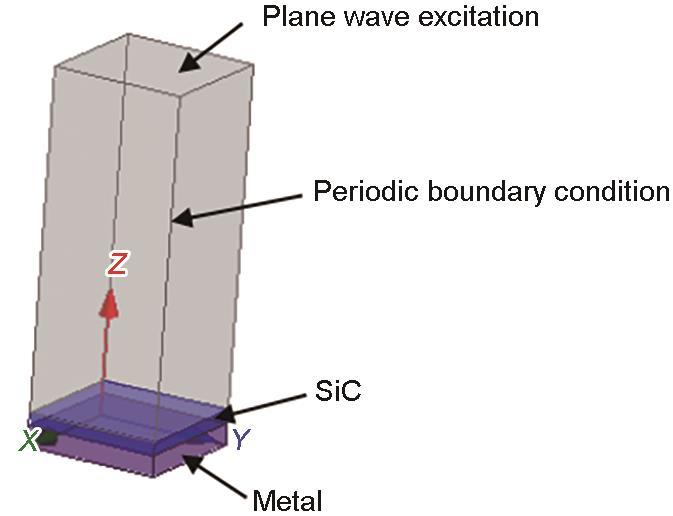
图9
SiC吸波材料反射率的仿真模型
Fig.9
Reflectivity simulation model of SiC absorbing material
图10
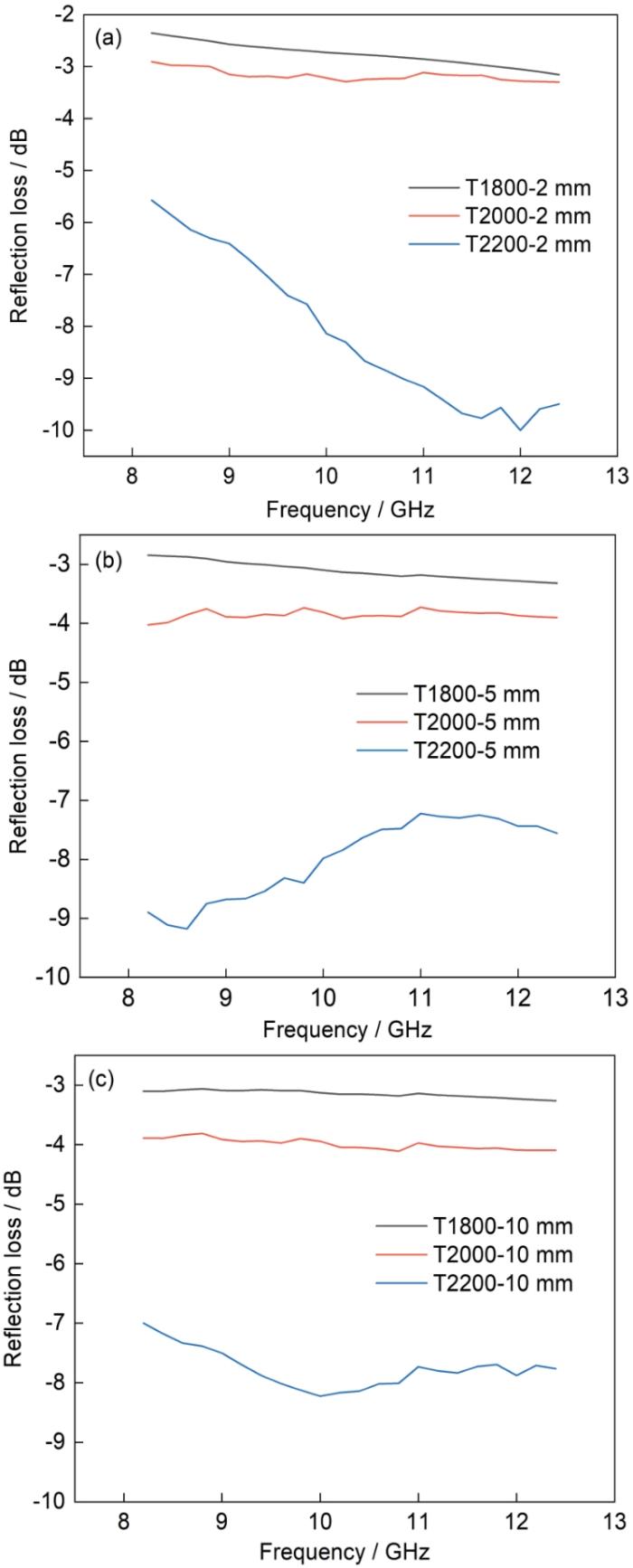
图10
不同厚度T1800、T2000、T2200试样的反射率
Fig.10
Reflectivity of T1800, T2000, T2200 specimens of different thicknesses
如图10所示,T1800试样在2 mm的反射率为-2dB至-3dB,表明烧结温度为1800℃制备的碳化硅其介电常数实部和虚部均较大,对电磁波的阻抗匹配恶化而发生强反射。T1800的厚度由2 mm增加到5 mm和10 mm,反射率均降低至约-3dB,虽然有一定的吸波能力但是对电磁波的反射仍然较多。T2000试样在三种厚度下的吸波性能都比T1800试样好。T2200试样在2 mm时反射率介于-5.5dB~-10dB,厚度增加到5 mm时反射率介于-7dB~-9.3dB,厚度为10 mm时反射率介于-7dB~-8.3dB。这表明,T2200试样的吸波性能明显优于其他两种试样。由图10可见,T1800和T2000的厚度增加后吸波性能略有提高。其原因是,虽然厚度增加是碳化硅的介电损耗能力提高,但是在部分波段,T2200试样的厚度增加却使其吸波性能降低。这与两次反射电磁波的谐振相关,表明T2200试样的介电损耗能力较弱,有一定的透波性[39]。
以上结果表明,虽然T1800试样的介电损耗能力最好,但是特征阻抗系数最低。阻抗匹配能力差产生电磁波强反射,不适合用做吸波材料。与T2200试样相比,T2000试样的介电损耗能力较强,阻抗匹配性能优于T1800试样,有一定的吸波能力;虽然T2200试样的介电损耗能力低于其他两种试样,但是其阻抗匹配性能最好,较多的电磁波进入其内被损耗,作为单层平板结构吸波性能最好。在整体上,T2000和T2200试样的介电损耗和阻抗匹配系数合适,适于用做吸波材料。依据使用要求,后续可基于两种SiC材料的不同电磁特性进行多层匹配或优化设计吸波结构。
3 结论
(1) 使热解碳与硅粉发生原位反应可制备SiC吸波材料。反应温度低于2000℃时生成3C结构的β相SiC,反应温度≥ 2000℃时不稳定的3C-SiC解离蒸发生成Si2C和SiC2气体,通过气相输运凝结在晶核上使晶粒快速变大,生成6H结构的α相SiC。随着反应烧结温度的进一步提高,SiC材料由β相加速向α相转变。
(2) 控制碳硅原位反应温度可调控生成的α-SiC的比例,从而影响SiC材料中的载流子浓度和材料的吸波性能。随着反应温度的进一步提高生成的SiC材料对电磁波的介电损耗能力逐步减弱,阻抗匹配性能逐步提高。反应温度≥ 2000℃时生成的SiC材料阻抗匹配和介电损耗性能适当,是较好的吸波材料。
参考文献
Research progress of MAX phase high temperature absorbing materials
[J].
MAX相高温吸波材料的研究展
[J].
High-temperature dielectric and microwave absorption properties of Si3N4-SiC/SiO2 composite ceramics
[J].
Research on micro-structure adjustment and microwave absorption properties of carbon-based composite materials
[D].
碳基复合材料的微结构调控及其电磁波吸收性能研究
[D].
Study on the properties of polysiloxane Pyrolysis CNTs/Si-O-C high temperature structural absorbing materials
[D].
聚硅氧烷热解CNTs/Si-O-C耐高温结构吸波材料性能研究
[D].
Research status and development trend of electromagnetic absorbing materials
[J].
电磁吸波材料的研究现状与发展趋势
[J].
Research progress of SiC composite microwave absorbing materials
[J].
SiC复合吸波材料的研究进展
[J].
Research progress of high temperature microwave absorption materials
[J].
耐高温吸波材料的研究进展
[J].
Preparation and electromagnetic wave absorption performance of Fe3Si/SiC@SiO2 nanocomposi-tes
[J].
Electromagnetic properties of coaxial core-shell CNTs@SiC prepared by chemical vapor deposition
[J].
化学气相沉积法制备同轴核壳结构CNTs@SiC的电磁特性研究
[J].
Development of continuously reinforced metal matrix composites for aerospace applications
[J].
Continuously reinforced metal matrix composites
[J].
Investigation on the influence of SiCf/TC17 composites preparation method on growth kinetics of interfacial reactive layer
[J].
SiCf/TC17复合材料制备方法对界面反应层生长动力学的影响
[J].
连续SiC纤维增强钛基复合材料(SiC<sub>f</sub>/Ti复合材料)具有良好的比强度和综合力学性能,是新一代装备研制备受关注的轻质高温结构材料。SiC<sub>f</sub>/Ti复合材料可采用箔压法(FFF)和基体涂层法(MCF)进行制备,为对比两种工艺方法对其界面反应生长的影响,采用FFF和MCF分别制备SiC<sub>f</sub>/TC17复合材料。对两种工艺制备的SiC<sub>f</sub>/TC17复合材料在高温下(800~900 ℃)进行热暴露处理,通过扫描电镜对其微观结构及界面反应层厚度进行分析,获得界面反应层在高温下的生长速率,并进一步获得不同制备工艺状态下材料的界面反应动力学参数。结果表明:相同温度下MCF法制备的SiC<sub>f</sub>/TC17复合材料界面反应速率大于FFF法制备的复合材料,前者的反应速率因子k<sub>0</sub>为4.942×10<sup>−3</sup> m/s<sup>1/2</sup>,反应激活能Q为276.3 kJ/mol,后者的界面反应速率因子k<sub>0</sub>为8.149×10<sup>−3</sup> m/s<sup>1/2</sup>,反应激活能Q为291.7 kJ/mol。这是由于MCF法制备的钛合金基体具有更微小的相组织,具有较小的反应激活能,在高温下具有更高的元素扩散速率。
Preparation of C/Zr0.5Hf0.5C-SiC composite by PIP process its microstructure and flexural proper-ties
[J].
PIP工艺制备C/Zr0.5Hf0.5C-SiC复合材料及其微观结构和弯曲性能
[J].基于自制Zr<sub>0.5</sub>Hf<sub>0.5</sub>C先驱体和商业化液态聚碳硅烷,通过先驱体浸渍裂解(PIP)工艺成功制备C/Zr<sub>0.5</sub>Hf<sub>0.5</sub>C-SiC复合材料,研究纤维表面热解C涂层厚度对复合材料微观结构及弯曲性能的影响。结果表明:自制Zr<sub>0.5</sub>Hf<sub>0.5</sub>C先驱体在1400 ℃下即可转化生成单一Zr<sub>0.5</sub>Hf<sub>0.5</sub>C固溶体。因具有良好的渗透性,转化生成的Zr<sub>0.5</sub>Hf<sub>0.5</sub>C基体同时存在于C/Zr<sub>0.5</sub>Hf<sub>0.5</sub>C-SiC复合材料的纤维束内和束间,呈包裹SiC基体的层状形貌。C/Zr<sub>0.5</sub>Hf<sub>0.5</sub>C-SiC复合材料主要由C,SiC和Zr<sub>0.5</sub>Hf<sub>0.5</sub>C相组成; 具有不同热解C涂层厚度(0.67,0.84,1.36 μm) 的3组复合材料密度分别为2.07,1.99,1.98 g/cm<sup>3</sup>; 随热解C涂层厚度的增加复合材料中SiC含量减少。弯曲加载中3组不同热解C涂层厚度复合材料均呈现假塑性断裂模式,弯曲强度,弯曲模量和断裂韧度分别在410 MPa,60 GPa和15.6 MPa·m<sup>1/2</sup>以上。良好的界面结合和预先引入的SiC基体是C/Zr<sub>0.5</sub>Hf<sub>0.5</sub>C-SiC复合材料获得优良弯曲性能的关键。
Preparation of SiC powders by carbonthermal reduction method at low temperature
[J].
碳热还原法低温制备碳化硅微粉
[J].以SiO2为硅源,炭黑为碳源,Fe2 O3为催化剂,采用碳热还原法在氩气保护下制备SiC微粉,研究催化剂含量,合成温度对SiC生成、形貌的影响.实验结果表明:在原料中添加Fe2 O3粉,1350℃保温3h就能产生SiC微粉;由X射线衍射分析显示,在1450℃下保温3h基本上全部转化为晶粒尺寸在50 nm左右SiC微粉;在相同温度下,随着Fe2 O3用量的增加,SiC产率增加.添加Fe2O3能加快反应速度以及提高SiC微粉的生成量.
New progress in preparation of high purity SiC micropowder
[J].The main preparation processes of high-purity SiC micropowder were reviewed,and the new progress of purification process of high-purity SiC micropowder in recent years was introduced.The upgrading of the preparation process,and the industrial technical process and equipment of high purity SiC micro-powder was prospected.
高纯SiC微粉制备进展
[J].综述了高纯SiC微粉主要制备工艺,介绍了近些年SiC微粉除杂提纯工艺新进展,提出未来高纯SiC微粉制备工艺应不断更新升级,产业化生产技术和装备也需要不断完善。
Research progress in preparation methods of SiC porous ceramics
[J].
SiC多孔陶瓷制备方法研究进展
[J].
Highly-porous hierarchical SiC structures obtained by filament printing and partial sintering
[J].
Fabrication and characterization of carbon fiber reinforced SiC ceramic matrix composites based on 3D printing technology
[J].
Research progress on silicon-based ceramic precursors for 3D printing
[J].
3D打印用硅基陶瓷前驱体研究进展
[J].
3D printing technology microwave absorption metamaterial absorbing honeycomb microwave absorbers ceramic matrix composites absorbers
[J].
3D打印微波吸收材料研究进展
[J].
B x O: Phases Present at High Pressure and Temperature
[J].
Coarsening of high purity SiC particles by gas phase transport
[J].
Reactions of silicon carbide and silicon (Ⅳ) oxide at elevated temperatures
[J].
Thermodynamic analysis of the high temperature stability of silicon nitride and silicon carbide
[J].
Effect of heat treatment temperature on microstructure and fracture strength of reaction-sintered silicon carbide
[J].
热处理温度对反应烧结碳化硅材料组织与性能的影响
[J].
Novel process for recrystallized silicon carbide through β-α phase transformation
[J].
Phase transformation during hot-pressing of cubic SiC
[J].
Thermodynamic analysis of phase transformations at the dissociative evaporation of silicon carbide polytypes
[J].
The influences of hot pressing on the microstructure and mechanical properties of SiC ceramic
[J].
热压工艺对SiC陶瓷结构及性能的影响
[J].
Corncob-derived hierarchical porous carbon/Ni composites for microwave absorbing application
[J]. J.
Progress in polymer-derived silicon carbide ceramics for microwave absorbing applications
[J].
聚碳硅烷转化碳化硅陶瓷吸波性能的研究进展
[J].
Self-healing superhydrophobic polyvinylidene fluoride/Fe3O4@polypyrrole fiber with core-sheath structures for superior microwave absorption
[J].
Study on three kinds of XRD quantitative analysis methods
[J].
三种X射线物相定量分析方法对比研究
[J].
Transmission electron microscopy characterization of grain structure and nanoparticles of 15-15Ti ODS austenitic steel
[J].
15-15TiODS奥氏体钢晶粒组织与纳米粒子的透射电镜表征
[J].
Synthesis and characterization of silicon carbide and titanium carbide nanomaterials
[D].
碳化硅和碳化钛纳米材料的制备与表征
[D].
Preparation and performance of self-assembled carbon/epoxy composite microwave absorbing coating
[J].
自组装碳/环氧树脂复合吸波涂料的制备及性能
[J].
Research on the application of pyrolytic carbon-based foam structure to thermal vacuum absorbing box
[J].
热解碳基泡沫结构应用于热真空吸波箱技术研究
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}