


氟橡胶是一类在主碳链和侧碳链上含有氟原子的合成聚合物弹性体,氟原子与碳原子形成了具有高键能(485 kJ/mol)的C-F键,可屏蔽和保护聚合物的主链[1,2]。因此,氟弹性体具有优异的热稳定性,耐油性,高抗氧化能力以及耐化学性[3~5]。但是,高分子量和强极性也使其加工性能有限,在一定程度上使其在现代实际生产和使用中的应用受到限制[6]。为了解决这一问题,人们开发了低分子量氟橡胶,即液体氟橡胶。这种液体氟橡胶不仅具有优异的热稳定性和化学稳定性,还具有固体含氟弹性体不具备的流动性和可塑性,易于加工、成型和固化,可用于制造密封胶、填缝剂、胶黏剂、涂层以及各种形状复杂制品并显著提高了加工性能[7,8]。
目前,液体氟橡胶的合成方法,有聚合法[9,10]和氧化降解法[11]。聚合法的过程复杂,需使用具有特殊结构的引发剂和调聚剂,且反应条件苛刻和反应可控的难度较大。氧化降解法可将高分子量的固体氟橡胶降解为分子链两端带有羧基的液体氟橡胶,其工艺简单,反应易控。但是,用氧化降解法合成的液体端羧基氟橡胶(CTLF)链中含有多种序列结构的双键(C=C),影响其热稳定性、化学稳定性以及后续官能化反应的活性和可控制性[12]。李东翰等[13,14]在合成液体端羟基氟橡胶的过程中发现,LiAlH4等强还原剂将端羧基还原为端羟基且可消除链中C=C,但是使氟的含量和化学稳定性降低。研究发现,用氟化加成反应可消除CTLF链中的C=C和提高其氟含量,实现其高性能化。
鉴于此,本文构建以Selectflour为氟化试剂、TBAF为亲核试剂、NBS为亲电试剂的氟化加成反应体系,以氟橡胶为原料用一锅法合成高性能的SCTLF、阐明其性能与分子链结构的构效关系并揭示其加成反应机理。
1 实验方法
1.1 实验用原料和试剂
固体氟橡胶(CGF-300A):工业级;1-氯甲基-4-氟-1,4-二氮二环[2,2,2]辛烷双四氟硼酸盐(Selectfour):分析纯;四丁基氟化铵(TBAF):分析纯;N-溴代丁二酰亚胺(NBS):分析纯;四氢呋喃(THF):分析纯。
1.2 SCTLF的合成和固化
将50.00 g固体氟橡胶置于容积为500 mL的三口烧瓶中,加入150 mL的四氢呋喃使其溶解,在10 ℃加入3.75 g的BTEAC、25.5 g的KOH溶液(45%,质量分数)和30 g的H2O2溶液,将混合溶液搅拌使其反应10 h。加入浓盐酸调节溶液的pH值为2.5,调速为500 r/min,再依次加入Selectfluor、TBA和NBS试剂,其摩尔比为[C=C]∶[Selectfluor]∶[TBAF]∶[NBS] = 1.0∶2.0∶1.0∶0.5,在30 ℃反应10 h。反应结束后,向体系中加入碳酸钙溶液(3%,质量分数)将产物洗涤,沉降两次后将产物收集并将其真空干燥至恒重。
将10 g的SCTLF溶解于10 mL丙酮中,按摩尔比[COOH]∶[NCO] = 1.0∶1.2称取HDI三聚体并将其溶解在2 mL丙酮中。将二者混合均匀后放入鼓风干燥箱中除去溶剂,然后将混合溶液倒入已预热的模具中并放入真空干燥箱除去溶剂,在90 ℃烘箱中固化8 h后得到固化产物。
1.3 羧基含量和双键含量的测定
将0.75 g液体端羧基氟橡胶溶解于40 mL丙酮中,充分溶解后滴加0.1 mL浓度为10 g/L的溴百里香酚蓝指示剂,将其搅拌均匀后用0.05 mol/L的KOH/C2H5OH对其标定,滴定至溶液变为绿色时为滴定终点。则羧基含量为
式中V为试样消耗KOH/C2H5OH溶液的体积,单位为mL;C为KOH/C2H5OH标准滴定溶液的浓度,单位为mol/L;m为测试样品的质量,单位为g。
将液体端羧基氟橡胶和液体端乙烯基氟橡胶各0.5 g溶解在30 mL乙酸丁酯中,然后依次加入6 mL质量分数为2.5%的氯化碘/冰乙酸溶液、15 mL三氯三氟乙烷和5 mL质量分数为3%的乙酸汞/冰乙酸溶液,振荡均匀后避光静置4 h,再加入15 mL质量分数为10%的KI溶液,静置1 h后加入50 mL去离子水,振荡摇匀后用0.1 mol/L的Na2S2O3溶液进行滴定,待接近滴定终点时加入5 mL质量分数为0.25%的淀粉溶液,振荡摇匀后待溶液分层时继续滴定,滴定至溶液的蓝色全部消失即为终点,并与空白实验结果对比,则双键含量为
式中V1为空白试样消耗Na2S2O3溶液的体积,单位为mL;V2为试样消耗Na2S2O3溶液的体积,单位为mL;m为液体端羧基氟橡胶质量或液体端乙烯基氟橡胶质量的准确数值,单位为g。
1.4 性能表征
使用Nicolet is10型红外光谱扫描仪测试FT-IR以定性分析其结构,扫描范围为400~4000 cm-1,扫描次数16次,溶剂为丙酮。GPC测试:将7 mg样品加入2 mL色谱级四氢呋喃(THF)将其完全溶解,用PL-50型凝胶渗透色谱仪测定样品分子量及其分布,用Empower Pto处理GPC数据,测试条件为:聚苯乙烯标准样品,测试流速1.0 mL/min,测试温度30 ℃,参数k = 38.11,α = 0.645。
用AVANCE-Ⅲ-500MHz型核磁共振仪(500 MHz)进行1H-NMR测试,用氘代丙酮(C3D6O)为氘代溶剂,标准物为四甲基硅烷(TMS);采用AC-80型NMR分析仪(470M)测试19F-NMR,标准物质为一氟三氯甲烷(CFCl3)。
使用美国TA热分析仪进行DSC测试。测试条件:用氮气保护,温度范围为-100~30 ℃,升温速率为10 ℃/min,测试产物的玻璃化转变温度(Tg)。使用TA Instruments-Waters LLC热失重分析仪测试TGA,测试条件为:在氮气条件下,测试温度为30~600 ℃,升温速度为10 ℃/min,测试产物的热分解温度(Td)。
用DV2T-RV型Brookfield粘度仪测定动力粘度,按照GB/T 2794-2013在20~70 ℃测试,每个样品测定3次取结果的平均值。
用3365型材料万能拉力试验机并根据GB/T528-2009标准进行测试样品的拉伸性能。拉伸速率为500 mm/min,测试温度为25 ℃,每个样品至少测试5个样条取其结果的平均值。
2 结果和讨论
2.1 反应温度、反应时间和试剂用量对氟化加成反应的影响
设定反应时间为10 h、Selectflour和TBAF为亲核试剂、NBS为亲电试剂,考察了反应温度对产物结构的影响,结果列于表1。可以看出,随着反应温度的升高C=C含量呈降低趋势,反应温度为30 ℃时C=C含量最低为0.10 mmol/g,羧基含量为2.25% (质量分数),分子量为2410。这表明,氟化加成反应体系的选择性较好,反应体系温和可控。
表1 反应温度对产物结构的影响
Table 1
| Temperature / oC | PDI | COOH / % | C=C / mmol·g-1 | |
|---|---|---|---|---|
| - | 2620 | 1.80 | 2.45 | 0.30 |
| 10 | 2510 | 1.88 | 1.94 | 0.26 |
| 20 | 2460 | 1.93 | 2.23 | 0.18 |
| 30 | 2410 | 1.92 | 2.25 | 0.10 |
| 40 | 2430 | 1.92 | 2.23 | 0.12 |
| 50 | 2450 | 1.92 | 2.22 | 0.13 |
| 60 | 2380 | 2.05 | 2.21 | 0.11 |
控制反应温度为30 ℃,保持其他反应条件不变,考察了反应时间对产物结构的影响,结果列于表2。可以看出,反应时间为10 h时双键含量显著降低(达到0.10 mmol/g),羧基含量的变化不大。随着反应时间的增加C=C含量不变,TBAF和NBS与羧基发生了副反应使羧基含量明显降低。反应时间为10 h氟化加成反应完全,且对端羧基结构影响较小。
表2 反应时间对产物结构的影响
Table 2
| Time / h | PDI | COOH / % | C=C / mmol·g-1 | |
|---|---|---|---|---|
| - | 2620 | 1.80 | 2.45 | 0.30 |
| 2 | 2510 | 1.88 | 2.38 | 0.26 |
| 4 | 2470 | 1.93 | 2.33 | 0.18 |
| 6 | 2480 | 1.95 | 2.30 | 0.14 |
| 8 | 2430 | 1.93 | 2.27 | 0.12 |
| 10 | 2410 | 1.92 | 2.25 | 0.10 |
| 12 | 2450 | 2.11 | 2.12 | 0.10 |
控制反应温度为30 ℃、反应时间为10 h,保持其他反应条件不变,考察了Selectflour试剂用量对产物结构的影响,结果列于表3。可以看出,摩尔比[Selectfluor]∶[C=C]为2.0∶1.0时C=C含量最低(达到0.08 mmol/g)。这表明,摩尔比[Selectfluor]∶[C=C]为2.0∶1.0时Selectfluor试剂充分参与了氟化加成反应,Selectfluor作为高反应活性的氟化试剂可以提供氟正离子进行高效的氟化加成反应。同时,过多Selectflour的强氧化性使部分Selectfluor试剂与链中C=C双键发生副反应,结果使分子链断裂、降低分子量、使C=C含量略有提高。
表3 Selectfluor用量对产物结构的影响
Table 3
| [Selectfluor]: [C=C] | PDI | COOH / % | C=C / mmol·g-1 | |
|---|---|---|---|---|
| - | 2620 | 1.80 | 2.45 | 0.30 |
| 1.0:1.0 | 2430 | 1.88 | 2.38 | 0.14 |
| 2.0:1.0 | 2410 | 1.85 | 2.30 | 0.08 |
| 3.0:1.0 | 2420 | 1.90 | 2.30 | 0.11 |
| 4.0:1.0 | 2490 | 1.95 | 2.27 | 0.13 |
| 5.0:1.0 | 2440 | 1.92 | 2.23 | 0.11 |
表4 TBAF用量对产物结构的影响
Table 4
| [TBAF]: [C=C] | PDI | COOH / % | C=C / mmol·g-1 | |
|---|---|---|---|---|
| - | 2620 | 1.80 | 2.45 | 0.30 |
| 0.5:1.0 | 2420 | 1.82 | 2.36 | 0.12 |
| 1.0:1.0 | 2410 | 1.85 | 2.30 | 0.08 |
| 1.5:1.0 | 2380 | 1.93 | 2.11 | 0.11 |
| 2.0:1.0 | 2420 | 2.01 | 1.98 | 0.13 |
| 2.5:1.0 | 2480 | 2.40 | 1.86 | 0.13 |
表5 NBS用量对产物结构的影响
Table 5
| [NBS]: [C=C] | PDI | COOH / % | C=C / mmol·g-1 | |
|---|---|---|---|---|
| - | 2620 | 1.80 | 2.45 | 0.30 |
| 0.5/1.0 | 2410 | 1.85 | 2.30 | 0.08 |
| 1.0/1.0 | 2420 | 1.82 | 2.23 | 0.11 |
| 1.5/1.0 | 2300 | 1.93 | 2.20 | 0.14 |
| 2.0/1.0 | 2440 | 1.88 | 2.09 | 0.12 |
| 2.5/1.0 | 2490 | 2.07 | 2.02 | 0.13 |
2.2 SCTLF的结构
图1

图2给出了氟化加成反应前后CTLF和SCTLF的1H-NMR谱。如图2所示,反应前后CTLF分子链均在谱中((3.51~2.30) × 10-6)处出现了与-CH2CF2-结构对应的特征峰;(a) (1.55 × 10-6)处的质子化学位移为分子链中-CF=C(CF3)-CH2-结构特征峰;(b) (4.68 × 10-6)处的质子化学位移为分子链中-(CF3)C=CH-结构特征峰;(c) ((7.70~7.75) × 10-6)处的质子化学位移为分子链中-CH=CF-结构特征峰;(d) (3.62 × 10-6)处的质子化学位移为分子链中-CHF-CH(CF3)-CF2-结构特征峰;(e) (1.78 × 10-6)处的质子化学位移为分子链中-CF2-CH(CF3)-CF2-结构特征峰[25];从图2可见,a、b、c三处对应的C=C特征峰明显减弱,表明链中含氟C=C发生了氟化加成反应,且饱和化的同时在d、e处产生了新的结构特征峰,分别对应-CHF-CH(CF3)-CF2-和-CF2-CH(CF3)-CF2-结构。
图2
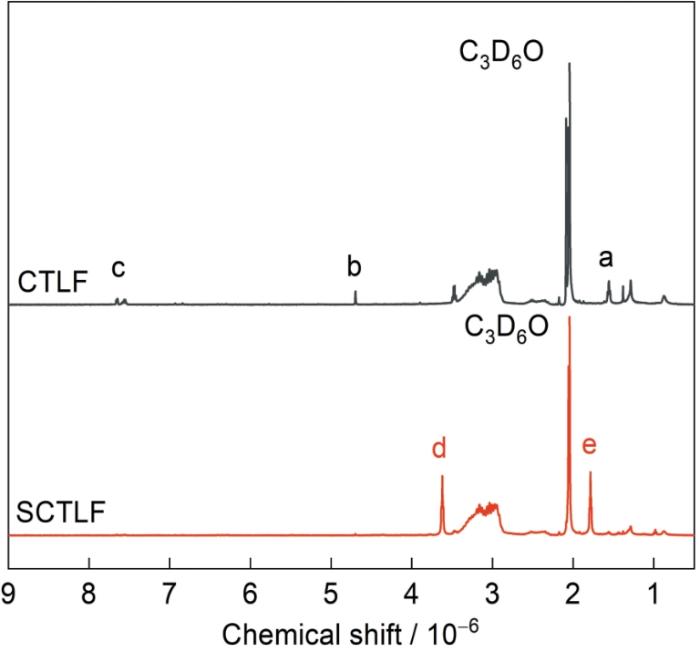
图3给出了氟化加成反应前后CTLF和SCTLF的19F-NMR谱。图3中c、e、f三处分别为分子链中所对应的含氟C=C结构特征峰,其中δ = -73.71(c)为-CF2CH=C(CF3)CF2-结构所对应的特征峰,符合Zaitsev消除规则;δ = -80.66(e)和δ = -81.30(f)分别为-CH=CFCF(CF3)-结构以及-CF=CHCF(CF3)CF2-结构所对应的特征峰,符合Hofmann消除规则。随着氟化加成反应的进行c、e、f三处的含氟C=C结构特征峰明显减弱,充分说明符合Zaitsev消除规则的含氟C=C反应活性更高,反应较完全,而符合Hofmann消除规则的含氟C=C在反应过程中发生的重排反应导致分子链中有部分双键残留[26~29]。特征峰的归属列于表6。
图3
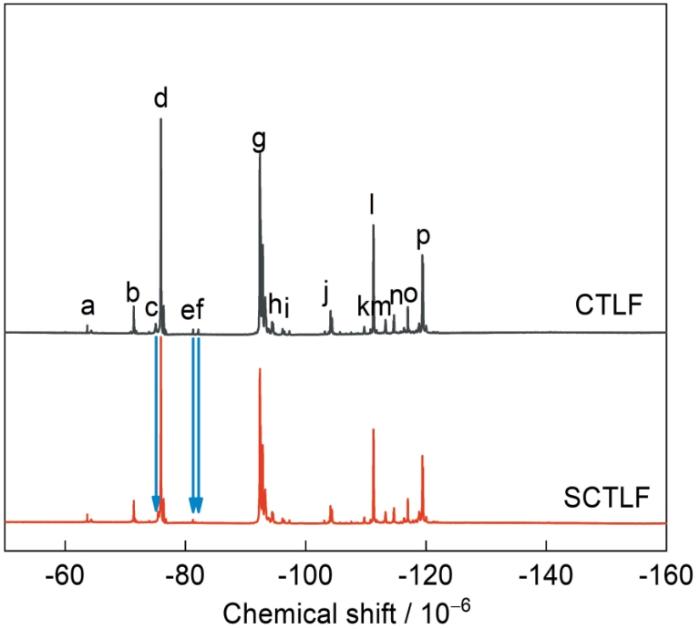
表6 CTLF和SCTF中19F-NMR谱中特征峰的归属
Table 6
| No. | δ / × 10-6 | Assignment |
|---|---|---|
| a | -63.46 | -CF2C |
| b | -70.67 | -CH2CF2CF(C |
| c | -73.71 | -CF2CH=C(C |
| d | -75.19 | -CF2CH2CF(C |
| e | -80.66 | -CH=CFCF(C |
| f | -81.30 | -CF=CHCF(C |
| g | -91.62 | -CF2CH2C |
| h | -93.56 | -CF2CH2C |
| i | -95.64 | -CH2CH2C |
| j | -103.62 | -CF2CH2C |
| k | -108.96 | -CF(CF3)CH2C |
| l | -110.51 | -CF2CH2C |
| m | -112.53 | -CF(CF3)CH2C |
| n | -113.95 | -CF2CH2C |
| o | -116.24 | -CH2CH2C |
| p | -118.72 | -CH2CF2C |
设定VDF序列结构对应特征峰面积为∑CF2 = I-63.46 + I-91.62 + I-93.56 + I-95.64 + I-103.62 + I-108.96 + I-110.51 + I-112.53 +I-113.95 + I-116.24;HFP序列结构对应特征峰面积为∑CF3 =I-70.67 + I-73.71 + I-75.19 + I-80.66 + I-81.30。于是,氟化加成反应前后CTLF中的单体含量(X)分别为
而样品中的氟含量(XF)则为
计算结果表明,氟化加成反应后SCTLF中的氟含量由原料的61.48%提高到65.47%。
图4

表7 CTLF和SCTLF的分子量和分子量分布
Table 7
| Type | PDI | |
|---|---|---|
| CTLF | 2620 | 1.80 |
| SCTLF | 2410 | 1.85 |
2.3 氟化加成反应的机理
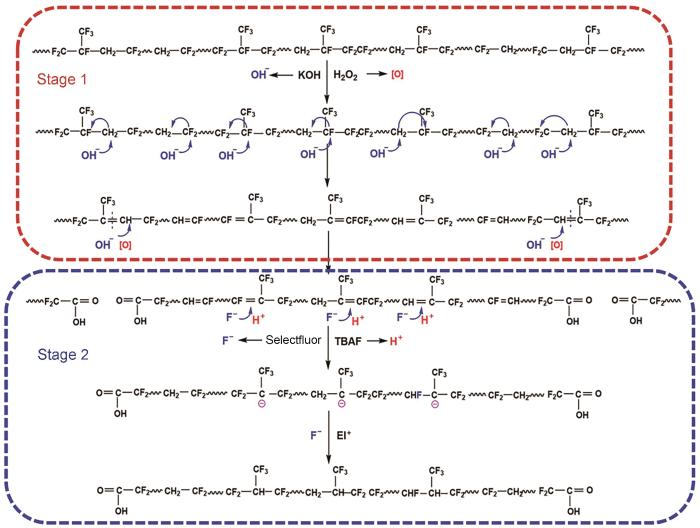
结构表征结果表明,针对CTLF创建的氟化加成反应体系可将其分子链中的C=C饱和化。氟化加成反应的机理,如图5所示。第一阶段:在碱性条件下氟橡胶先发生以Zaitsev消除规则为主、Hofmann消除规则为辅的脱氟加氢反应和双键重排反应,在链中生成C=C,随后发生氧化降解反应使部分C=C氧化断键,在分子链端生成端羧基[11,12]。第二阶段:Selectflour和TBAF协同发生亲核加成反应在分子链中的C=C中引入氟离子形成C-F键,反应过程遵循马施规则。然后,在NBS与链中剩余的C=C发生亲电加成反应的过程中,F离子的电负性大于Br离子使Selectfluor中的F离子进一步发生加成反应并继续生成C-F键,使链中的双键饱和化。
图5
2.4 SCTLF的热学性能
图6

图7给出了对氟化加成反应前后CTLF的Td的测定结果。与CTLF相比,SCTLF的初始热分解温度T5%由243升高到270 ℃,Tmax由448 ℃升高至458 ℃。其原因是,随着氟化加成反应的进行分子链中的含氟C=C被饱和化而使分子链柔顺性降低和刚性增强。同时,氟原子的引入显著提高了聚合物中C-F键的稳定性,在提高其热分解温度和抗氧化性的同时减少侧链反应和形成物理障碍,从而提高了聚合物的整体热稳定性。这进一步证明,分子链发生了氟化加成反应,与反应机理相符。
图7
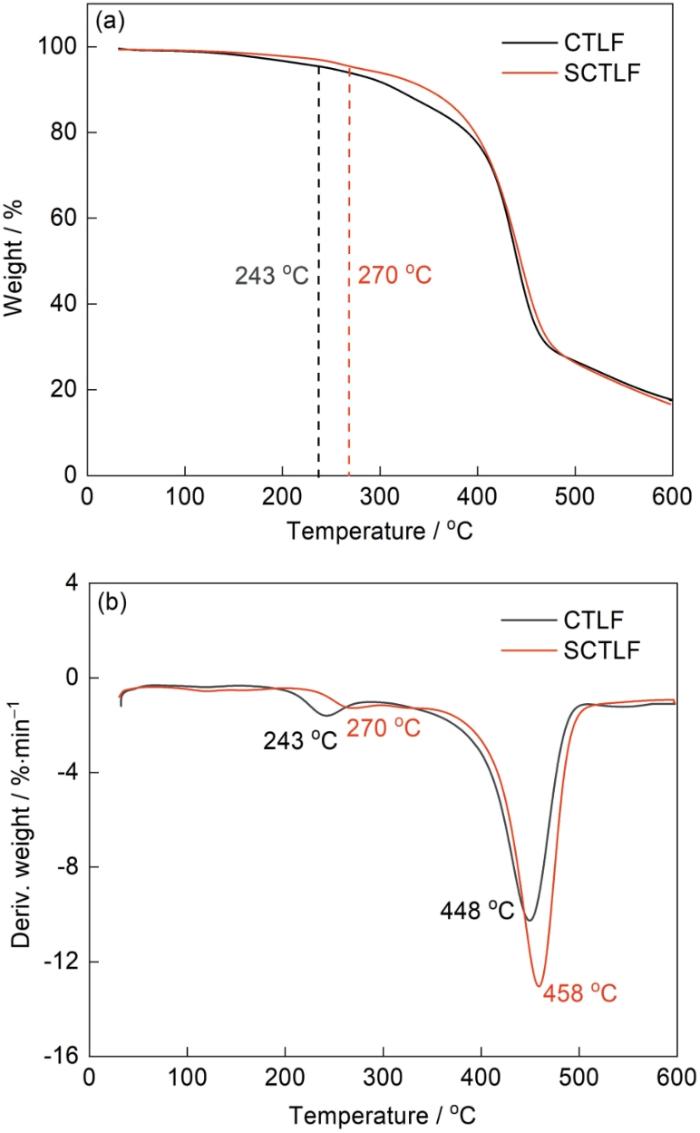
图7
CTLF和SCTLF的TGA和DTG谱
Fig.7
TGA (a) and DTG (b) thermograms of CTLF and SCTLF
2.5 SCTLF的动力粘度
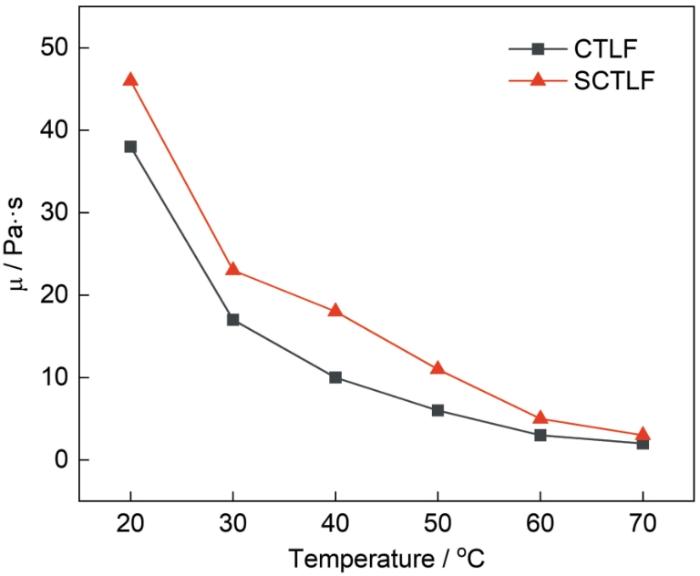
图8表明,CTLF和SCTLF的动力粘度随测试温度的提高而降低。测试温度为20 ℃~50 ℃动力粘度随着温度的提高迅速降低,测试温度高于50 ℃后动力粘度随着温度的提高下降趋势缓慢,测试温度70 ℃时CTLF和SCTLF的动力粘度分别降低为2 Pa·s和3 Pa·s。剪切速率不变时,SCTLF的动力粘度主要取决于自由体积和大分子之间的缠结。测试温度越高,分子内的自由体积越大则分子间相对滑动较为容易,使CTLF和SCTLF的流动性能更好,动力粘度降低。氟化加成反应消除了SCTLF链中的不饱和双键,使分子链柔顺性降低和刚性提高,更多极性较强的C-F键也使分子间作用力增强,阻碍了分子链的相对运动。因此,与CTLF相比,在相同温度下SCTLF的动力粘度较大。
图8
2.6 SCTLF的固化
图9
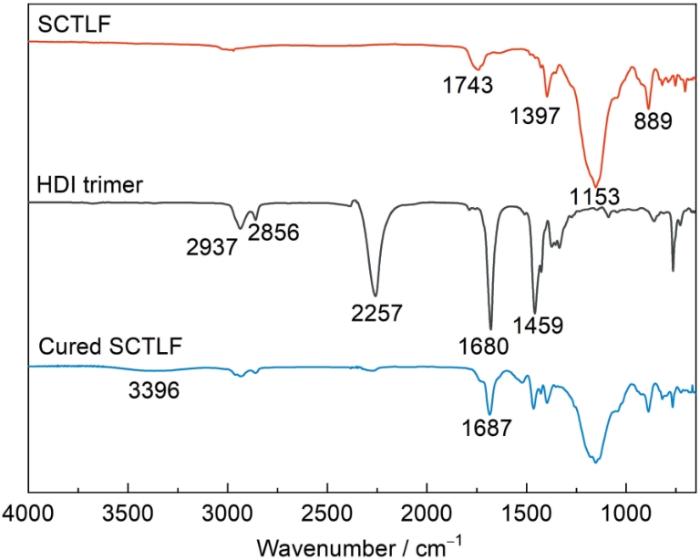
图9
SCTLF、HDI三聚体以及固化产物的FT-IR谱
Fig.9
FT-IR spectra of SCTLF, HDI trimer and cured SCTLF
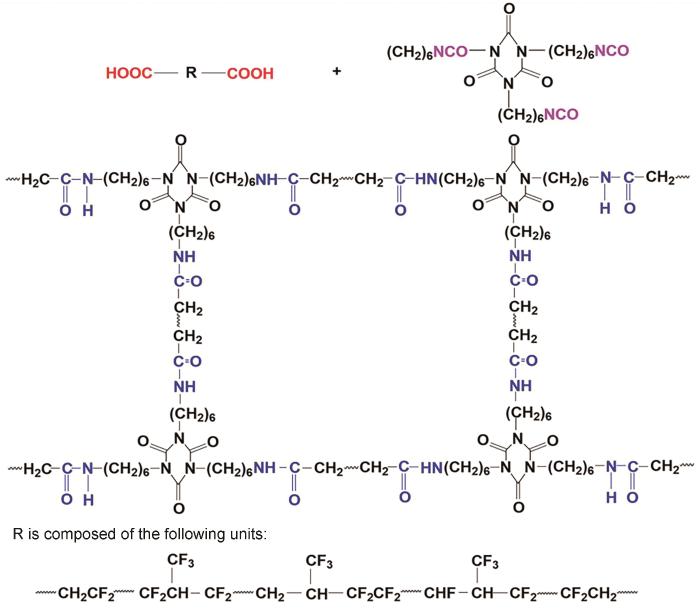
图10 给出了SCTLF与HDI三聚体的固化机理。根据异氰酸酯基团的电子共振理论,NCO基团的共振作用使其电荷分布不均:氮氧原子成了亲核中心,碳原子成了亲电中心。因此,NCO基团极易与羧基发生反应。在固化反应过程中,-NCO先与-COOH反应生成热稳定性较低的羧基酸酐,其在高温下分解生成酰胺键和CO2,从而形成交联网络结构而实现了SCTLF的固化。
图10
HDI三聚体分别使CTLF和SCTLF固化,摩尔比[COOH]∶[NCO]=1.0∶1.5时固化产物的力学性能,在图11中给出。氟化加成反应后,SCTLF链中C=C的含量降低而分子链刚性增强,SCTLF固化产物具有更加优异的力学性能:拉伸强度为1.49 MPa,断裂伸长率为140%。
图11

图11
CTLF固化产物和SCTLF固化产物的力学性能
Fig.11
Mechanical properties of cured CTLF and cured SCTLF
测试CTLF和SCTLF固化产物的溶胀度,以研究其耐航空煤油和耐酸碱性能,结果如图12所示。结果表明,在这几种介质中,CTLF和SCTLF的固化产物浸泡24.0 h和72.0 h后其质量变化率均小于5.00%,表明其化学稳定性良好。同时,氟化加成反应消除了SCTLF链中的C=C,使其化学稳定性更加优异。
图12
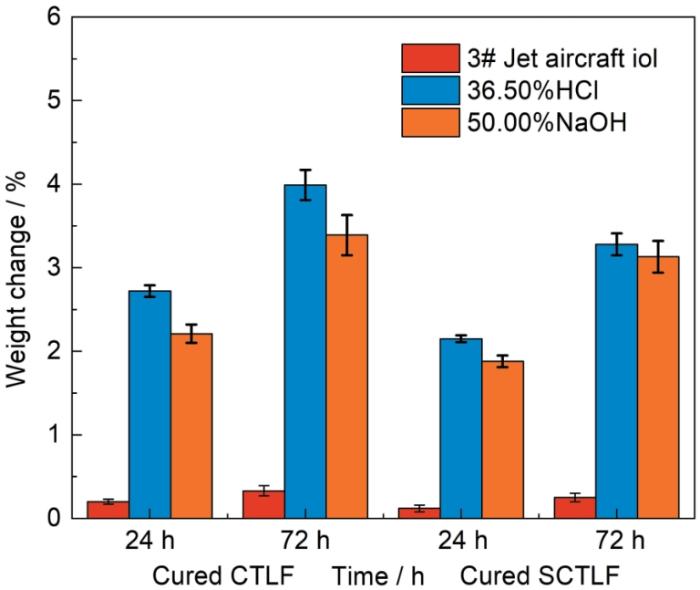
图12
CTLF固化产物和SCTLF固化产物的耐航空煤油和耐酸碱性能
Fig.12
Aircraft oil, acid and alkali resistance of cured CTLF and cured SCTLF
3 结论
以氟橡胶为原料,用氟化加成体系可制备SCTLF,在消除链中不饱和双键的同时提高了氟含量。符合Zaitsev消除规则产生的C=C活性较高,加成反应完全;因发生重排反应,符合Hofmann消除规则产生的C=C有部分残留。以HDI三聚体为固化剂可实现SCTLF的固化。这种固化产物具有优异的化学稳定性。
参考文献
Synthesis and properties of the novel high-performance 2 hydroxyl-terminated liquid fluoroelastomer
[J].
Reduction of liquid terminated-carboxyl fluoroelastomers using NaBH4/SmCl3
[J].
Investigation of the structure and piezoelectricity of poly (vinylidene fluoride-trifluroethylene) copolymer doped with different dyes
[J].
Structure and thermal stability of fluororubber
[J].
氟橡胶结构及热稳定性研究
[J].
Durability and service life prediction of fluorocarbon elastomer under thermal environments
[J].
Atom transfer radical polymerization initiated with vinylidene fluoride telomers
[J].
Chemical degradation of an uncrosslinked pure fluororubber in an alkaline environment
[J].
Photochemical induced polymerization of vinylidene fluoride (VDF) with hydrogen peroxide to obtain original telechelic PVDF
[J].
Photochemically cross-linked perfluoropolyether-based elastomers: synthesis, physical characterization, and biofouling evaluation
[J].
Iodine transfer terpolymerization of vinylidene fluoride, α-trifluoromethacrylic acid and hexafluoropropylene for exceptional thermostable fluoropolymers/silica nanocomposites
[J].
Study on the dehydrofluorination of vinylidene fluoride (VDF) and hexafluoropropylene (HFP) copolymer
[J].
Preparation of telechelic hydroxyl low molecular weight fluoropolymers
[J].
Preparation of low molecular weight fluoropolymer with hydroxyl end by borohydride/rare earth chloride reduction system
[P]. China, CN107674133B, 2019
硼氢化物/稀土元素氯化物还原体系制备端羟基低分子量含氟聚合物的方法
[P]. 中国, CN107674133B, 2019
Straightforward access to N-trifluoromethyl amides, carbamates, thiocarbamates and ureas
[J].
Research progress of fluorinated reagents and application of selectfluor (F-TEDA-BF4) in organic synthesis
[J].
氟化试剂的研究进展及Selectfluor在有机合成中的应用
[J].
Recent advances in the application of selectfluor as a “fluorine-free” functional reagent in organic synthesis
[J].
A new method for the synthesis of fluoro-carbohydrates and glycosides using selectfluor
[J].
DBFOX-Ph/metal complexes: Evaluation as catalysts for enantioselective fluorination of 3-(2-arylacetyl)-2-thiazolidinones
[J].
Anhydrous tetrabutylammonium fluoride
[J].
Tetrabutylammonium fluoride (TBAF) is prepared at low temperature by nucleophilic aromatic substitution of hexafluorobenzene with tetrabutylammonium cyanide. Adventitious water is scavenged during this synthesis by the generated hexacyanobenzene, which readily adds water under basic conditions. Contrary to expectations, TBAF is stable to Hofmann elimination in polar aprotic solvents under anhydrous conditions. Added hydroxylic solvents are shown to catalyze the decomposition of TBAF and to catalyze proton exchange with DMSO. The synthetic utility of this salt is described briefly.
Operationally simple and efficient workup procedure for TBAF-mediated desilylation: application to halichondrin synthesis
[J].An operationally simple and efficient workup method for tetrabutylammonium fluoride (TBAF)-mediated t-butyldimethylsilyl (TBS) deprotection has been developed. The procedure includes addition of a sulfonic acid resin and calcium carbonate, followed by filtration and evaporation. This method eliminates the tedious aqueous-phase extraction process to remove excess TBAF and materials derived from TBAF, thereby making the protocol highly amenable to multiple TBS deprotections. Its efficiency and usefulness were demonstrated by using the transformation of 1a to 3a in the halichondrin synthesis. [reaction: see text].
A convenient preparation of 5-iodo-1, 4-disubstituted-1, 2, 3-triazole: Multicomponent one-pot Reaction of azide and alkyne mediated by CuI-NBS
[J].
Structure, preparation and properties of liquid fluoroelastomers with different end groups
[J].In order to design and prepare liquid fluoroelastomers with different end groups, and reveal the relationship between the molecular chain structure and properties, we studied on the oxidation degradation method and functional group conversion method to prepare carboxyl-terminated and hydroxyl-terminated liquid fluoroelastomers, respectively. The reaction mechanisms were also deduced. Furthermore, the curing system was created for liquid fluoroelastomers, and systematically analyzed their properties. The sequence type and content of the -C[double bond, length as m-dash]C- and oxygen-containing groups in the samples were measured and characterized by attenuated total reflectance/Fourier transform infrared (ATR-FTIR) spectroscopy, H nuclear magnetic resonance (H-NMR), F-NMR spectroscopy and chemical titration, the molecular weights of liquid fluoroelastomers were measured by gel permeation chromatography (GPC). Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were used to examine the thermal properties, while a viscometer was used to measure the dynamic viscosity of the liquid fluoroelastomers. Then the mechanical and surface properties of the cured samples were examined by universal testing machine and contact angle measurement instrument, respectively. The results show that carboxyl-terminated liquid fluoroelastomer with 2.71 wt% carboxyl terminal groups can be prepared by oxidation degradation method. When lithium aluminium hydride (LiAlH) was used as the reducing agent, it can efficiently convert carboxyl group to hydroxyl group with a conversion rate of more than 95%. In addition, it can be seen that the dynamic viscosity of the liquid fluoroelastomers were all decreased with the increase of temperature, and it is similar to about 10 Pa s at 70 °C. Compared with carboxyl-terminated liquid fluoroelastomers, hydroxyl-terminated liquid fluoroelastomers has higher curing reactivity, higher glass transition temperature () and thermal decomposition temperature (), and better mechanical properties of cured samples. The two types of liquid fluoroelastomers with distinct end groups presented distinct hydrophilicity.This journal is © The Royal Society of Chemistry.
Spectrometric identification of organic compounds
[J].
Dehydrofluorination mechanisms and structures of fluoroelastomers in alkaline environments
[J].
Use of bis (trifluoromethyl) peroxy dicarbonate as initiator in the radical homopolymerisation of vinylidene fluoride (VDF) and copolymerisation of VDF with hexafluoropropylene
[J].
Fluorinated cotelomers based on vinylidene fluoride (VDF) and hexafluoropropene (HFP): Synthesis, dehydrofluorination and grafting by amine containing an aromatic ring
[J].
Addition reaction of halogens to vinyl (pentafluorocyclopropanes): Competition between a radical addition and an electrophilic addition
[J].
2, 2-Dihydroperfluoropentane (HFC 4310 mf) synthesis from HFP dimer
[J].
C-F activation reactions at germylium ions: dehydrofluorination of fluoralkanes
[J].
Synthesis and characterizations of novel proton-conducting fluoropolymer electrolyte membranes based on poly (vinylidene fluoride-ter-hexafluoropropylene-ter-α-trifluoromethacrylic acid) terpolymers grafted by aryl sulfonic acids
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}