


作为一种新型热固性树脂,苯并噁嗪具有良好的热性能、机械性能、耐化学腐蚀性和灵活的分子设计性、固化时近零的体积收缩、低表面能和吸水率等特点,在摩擦材料、航空航天、电子等领域得到了广泛的应用。但是苯并噁嗪的固化温度较高、固化速率较低,限制其在很多领域的应用[1,2,3,4,5,6]。噁嗪环的扭曲半椅式结构较不稳定,受热时环上的C-O键断裂,产生碳正离子中间体并进一步引发聚合反应。噁嗪环Ar-O-C上C-O键中的O或C原子与外加催化剂(包括质子酸、路易斯酸、酚和胺等)中的-OH、过渡金属离子或富电子结构等形成的氢键等相互作用,会促进C-O的断裂、降低苯并噁嗪的开环聚合温度[7,8,9,10,11,12,13,14]。但是,这些外加催化剂,包括有机质子酸、路易斯酸和小分子胺类等,其使用条件苛刻、毒性较大或在高温较易挥发。聚酰胺650是一种常用的环氧树脂固化剂,几乎无毒、无挥发性,分子结构中含有酰胺基结构和较长的脂肪碳链,因此还可作为一种增韧剂来改善环氧树脂的脆性[15]。酰胺结构中的-NH或富电子的O、N原子与噁嗪环上的O或N原子形成氢键或其他作用,影响苯并噁嗪的开环聚合。同时,苯并噁嗪聚合后-CONH-中的O、N及H原子也可能与其酚羟基上的H以及Mannich(-CH2-NR-CH2-)桥上的N原子形成氢键,改变聚苯并噁嗪的物理交联点、影响其性能。此外,O-PA中的脂肪链也会提高聚苯并噁嗪的韧性。本文使用O-PA作为苯并噁嗪开环聚合催化剂促进开环聚合和增韧改性,研究其对聚苯并噁嗪热稳定性的影响。
1 实验方法
1.1 实验用材料
实验用材料有:多聚甲醛、4,4’-二氨基二苯甲烷;双酚A、苯胺;苯酚;甲苯、丙酮、乙醇;聚酰胺650(O-PA)。
用溶剂法制备双酚A/苯胺型苯并噁嗪(Ba)、苯酚/苯胺型苯并噁嗪(Pa)和苯酚/4,4’-二氨基二苯甲烷型苯并噁嗪(Pddm)。
1.2 聚酰胺650与苯并噁嗪混合体系的制备
O-PA/Ba混合体系的制备:将O-PA、Ba和丙酮分别加入烧瓶后在50℃搅拌1 h,然后用旋转蒸发仪和真空干燥脱除溶剂,得到O-PA/Ba混合体系。其中O-PA与Ba的质量比为0:100、5:95、10:90、15:85、20:80,分别记做Ba、5Ba、10Ba、15Ba和20Ba。
O-PA/Pa和O-PA/Pddm的制备过程与O-PA/Ba的相同,O-PA与苯并噁嗪(Pa或Pddm)的质量比为0:100、5:95、10:90、15:85、20:80,分别记做Pa、5Pa、10Pa、15Pa、20Pa和Pddm、5Pddm、10Pddm、15Pddm、20Pddm。
1.3 聚苯并噁嗪的制备
将苯并噁嗪或O-PA/苯并噁嗪混合体系置于自制的铝制模具中,再将其放在真空干燥箱中一定温度下抽至无气泡产生;然后转移至鼓风干燥箱中分阶段固化。
O-PA/Ba固化物的制备:在110℃抽真空后在鼓风干燥箱中,按程序120℃/1 h,130℃/1 h,140℃/1 h,150℃/1 h,160℃/1 h,170℃/2 h,190℃/4 h进行固化。将Ba及其混合体系的固化物分别记做:PBa、P5Ba、P10Ba、P15Ba和P20Ba。
O-PA/Pa固化物的制备:在110℃抽真空,接着在鼓风干燥箱中按步骤120℃/1 h,130℃/1 h,140℃/1 h,150℃/1 h,160℃/1 h,170℃/1 h,180℃/1 h,190℃/2 h固化。Pa及其混合体系的固化物分别记做PPa、P5Pa、P10Pa、P15Pa、P20Pa。
O-PA/Pddm固化物的制备:在120℃抽真空后转至鼓风干燥箱中按步骤130℃/1 h,140℃/1 h,150℃/1 h,160℃/1 h,170℃/5 h,180℃/3 h,200℃/1 h固化。Pddm及其混合体系的固化物分别记做PPddm、P5Pddm、P10Pddm、P15Pddm、P20Pddm。
1.4 性能表征
使用GT-Ⅲ型凝胶化时间测试仪测试凝胶化时间。分别在160℃、170℃和180℃测试Ba、Pddm和Pa的凝胶化时间。温度稳定后将约20 mg的样品放入盘中,不断搅拌和挑起样品直到无法拉丝时为止。
使用IS 10型红外光谱仪测试傅里叶变换红外光谱(FT-IR)。用KBr压片法制备样品,扫描范围为4000~400 cm-1。使用TA Q20差示扫描量热仪进行差示扫描量热分析(DSC)。在氮气气氛中进行,流量50 ml/min,升温速率分别为:2、5、10、15和20℃/min,测试范围为40~320℃。使用 Discovery系列进行热重分析(TGA)。在氮气气氛中进行,流量为25 ml/min,升温速率10℃/min,测试范围40~810℃。按照GB/T 1043.1-2008,使用XJJD-5.5型电子简支梁冲击试验机测量冲击性能,样条的尺寸为80×10×4 mm3,冲头规格2 J,取5次实验结果的平均值。
2 结果和讨论
2.1 聚酰胺650对苯并噁嗪开环聚合的影响
表1 O-PA对Ba(160℃)、Pddm(170℃)和Pa(180℃)凝胶化时间的影响
Table 1
| Sample | Ba | 5Ba | 10Ba | 15Ba | 20Ba | Pa | 5Pa | 10Pa | 15Pa | 20Pa | Pddm | 5Pddm | 10Pddm | 15Pddm | 20Pddm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tgel/s | 5000 | 1409 | 677 | 469 | 500 | 1043 | 489 | 312 | 206 | 186 | 1410 | 307 | 198 | 150 | 119 |
为进一步说明O-PA对苯并噁嗪开环聚合的影响,进行了O-PA/苯并噁嗪混合体系的DSC测试,结果如图1和表2所示。可以看出,随着O-PA的引入苯并噁嗪开环聚合起始温度(Ti)和峰值温度(Tp)均向低温处移动,进一步说明O-PA对苯并噁嗪有明显的催化作用,与凝胶化时间的测试结果一致。但是,除Pa外,当O-PA的添加量超过5%后Tp的变化较小,只是Ti随O-PA的增加而不断降低。其原因是,O-PA的量越多则-CONH-与噁嗪环上O、C原子形成氢键等相互作用的机会越多(图2),使噁嗪环更易开环。此外,表2还表明,当O-PA的添加量为5%时O-PA/苯并噁嗪混合体系的聚合热焓(ΔH)最大,分别超过Pa、Ba和Pddm的聚合热焓。这意味着,O-PA的引入使苯并噁嗪的聚合程度提高。进一步增加O-PA的添加量,混合体系的ΔH有所下降。其原因,一方面是混合体系中苯并噁嗪的量不断减少,使聚合热降低; 还有可能是在低温搅拌及干燥过程中已有部分苯并噁嗪发生了开环聚合反应,且O-PA的量越多低温搅拌及干燥过程中发生开环聚合的程度越高,使其热焓越小。另外,图1也表明,O-PA的添加使苯并噁嗪聚合放热峰明显变宽,减弱了反应放热的集中,这有利于苯并噁嗪的加工成型。
图1
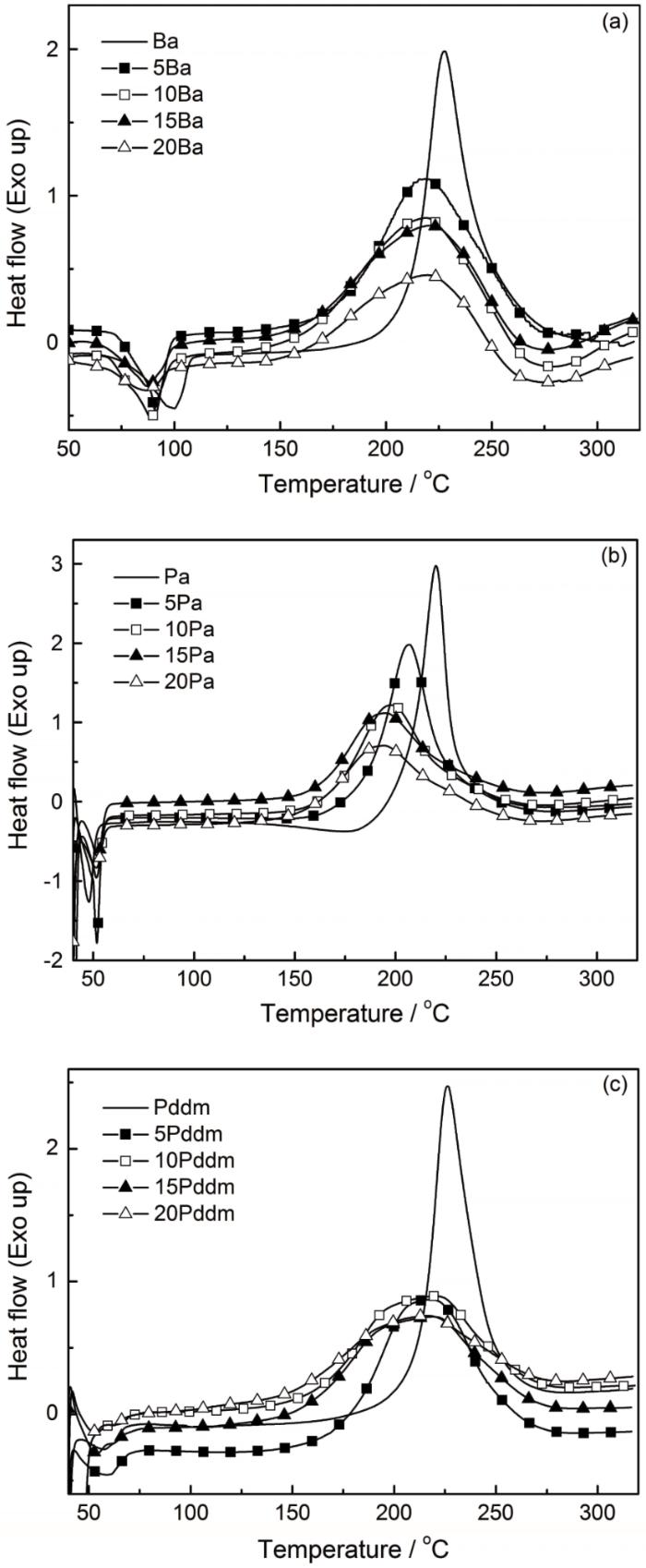
图1
O-PA/苯并噁嗪的DSC曲线
Fig.1
DSC curves of O-PA/benzoxazine (a) O-PA/Ba; (b) O-PA/Pa; (c) O-PA/Pddm
表2 O-PA/Pa、O-PA/Ba和O-PA/Pddm的DSC测试结果
Table 2
| Ba | 5Ba | 10Ba | 15Ba | 20Ba | Pa | 5Pa | 10Pa | 15Pa | 20Pa | Pddm | 5Pddm | 10Pddm | 15Pddm | 20Pddm | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ti/℃ | 156.8 | 107.1 | 104.9 | 104.8 | 98.3 | 177.0 | 149.0 | 143.1 | 136.3 | 127.8 | 178.3 | 133.8 | 116.4 | 92.4 | 69.5 |
| Tp/℃ | 227.8 | 218.7 | 218.9 | 221.0 | 219.7 | 220.1 | 207.1 | 198.8 | 195.4 | 194.2 | 226.3 | 217.1 | 214.0 | 218.0 | 214.6 |
| ΔH/J·g-1 | 342.7 | 361.4 | 388.3 | 328.0 | 284.3 | 373.1 | 417.1 | 343.8 | 300.9 | 304.7 | 347.4 | 363.6 | 311.6 | 338.2 | 300.8 |
图2
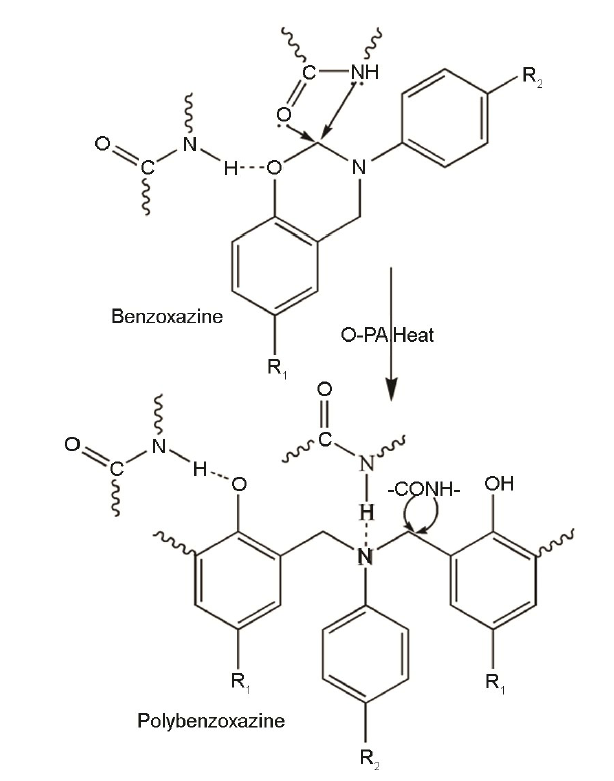
图2
O-PA与苯并噁嗪及其聚合物间的相互作用
Fig.2
Interaction between O-PA and benzoxazine or polybenzoxazine
以Ba和10Ba为例,进行了FTIR测试。图3给出了Ba和10Ba在50℃搅拌1 h和不同固化阶段后的FTIR图。苯并噁嗪开环后形成一对可逆平衡的亚胺离子(CH2=N+<)和碳正离子中间体,其中1652和1649 cm-1分别为Ba和10Ba中亚胺离子中间体的吸收峰;1230和1025 cm-1、1233和1030 cm-1分别为Ba、10Ba中C-O-C的不对称和对称伸缩振动吸收峰;950 cm-1处为噁嗪环的特征峰。
图3
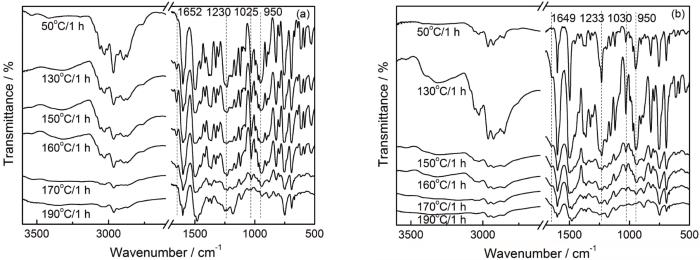
图3
Ba和10Ba在不同固化阶段的FTIR图
Fig.3
FTIR spectra of Ba (a) and 10Ba (b) at the different cured stages
对于Ba,随着固化温度的提高其在1652 cm-1处的吸收峰表现出出现-增强-减弱-消失的趋势。这意味着,噁嗪环开环生成亚胺离子中间体,且随温度升高、噁嗪环开环程度的提高亚胺离子增加;碳正离子中间体与酚羟基的邻位(或芳胺的对位)发生亲电取代反应,使亚胺离子减少、直至消失[16]。此外,Ba中噁嗪环及噁嗪环上的C-O-C吸收峰也在逐渐减弱。在170℃固化1 h后这些吸收峰显著减弱,但是直到在190℃固化1 h这些特征峰仍明显可见。
对于10Ba体系,其亚胺离子吸收峰、噁嗪环特征峰的变化趋势与Ba相似。但是与Ba明显不同的是,在150℃固化1 h后噁嗪环及其C-O-C的吸收峰明显减弱;在190℃固化1 h后这些特征峰几乎消失。上述现象进一步说明,O-PA能催化苯并噁嗪的开环聚合。但是峰的位置和形状表明,Ba和10Ba的情况基本相同,意味着O-PA的添加不影响苯并噁嗪聚合过程中的化学结构转变。
2.2 聚酰胺650对苯并噁嗪开环聚合动力学的影响
在动力学研究中,常用Kissinger和Ozawa 方法计算反应活化能,其方程为
Kissinger法:
Ozawa法:
式中F(α)是常数方程;β=dT/dt是升温速率;Tp为峰值温度;Ea是反应活化能;R是气体常数(8.314 J/(mol·K));A是指前因子。对Ba和10Ba体系进行非等温DSC测试,并用Kissinger和Ozawa方法计算其开环聚合反应的表观活化能(Ea),结果如图4所示。
图4
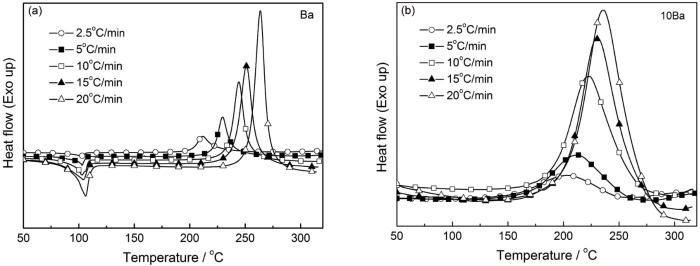
图4
Ba和10Ba的DSC曲线
Fig.4
DSC curves of Ba (a) and 10Ba (b) for the different heating rate
对于Kissinger方法,以ln(β/Tp2)、1/T分别为纵、横坐标作图;对于Ozawa方法,以lnβ、1/T分别为纵、横作图,所得拟合直线如图5所示。根据拟合直线的斜率可分别计算出Ea。其中,用Kissinger方法所得Ba和10Ba的Ea分别为82.46和119.66 kJ/mol,用Ozawa方法所得Ba和10Ba的Ea分别是86.43和121.05 kJ/mol。通常Ea值越小、反应越容易进行,但是聚酰胺650的添加使Ba开环聚合反应活化能明显增大,即Ea的测试结果与凝胶化时间和DSC测试结果相反。以前的研究结果也表明,磷酸锆等催化剂的引入在降低苯并噁嗪开环聚合温度的同时也使其Ea增大[12,17,18],因为磷酸锆抑制苯并噁嗪链段的运动。O-PA使Ba开环聚合反应活化能增加,因为苯并噁嗪链段的运动受到了抑制,使固化反应需要克服更高的能垒。因为O-PA中酰胺键上的O、H原子分别与Ba开环后形成的酚-OH和Mannich桥上的N原子形成氢键(图2),抑制了Ba中链段的移动。同时,随着开环聚合的进行和酚羟基等的增加,这种抑制作用不断增大,导致Ba的Ea增大。
图5
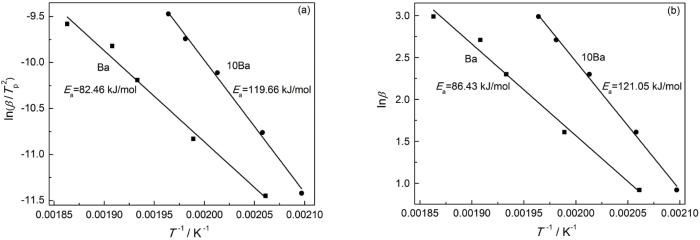
图5
Ba和10Ba的Kissinger和Ozawa拟合曲线
Fig.5
Kissinger (a) and Ozawa (b) fitting curves of Ba and 10Ba
2.3 聚酰胺650对聚苯并噁嗪化学结构的影响
以Ba为例研究了O-PA对苯并噁嗪固化物化学结构的影响。PBa和O-PA/Ba固化体系的FTIR测试结果,如图6所示。其中3600~3120 cm-1之间为PBa中酚-OH及O-PA中酰胺键上>NH的伸缩振动吸收峰,1706~1646 cm-1之间的肩峰(PBa无此峰)为O-PA中C=O吸收峰,1617~1447 cm-1之间是苯环骨架振动吸收峰和O-PA中N-H弯曲振动吸收峰。由于PBa中的-OH和N与O-PA中的C=O和>NH之间可以形成各种形式的氢键(图2),而固化体系中O-PA的含量不同,使各固化体系在3600~3120 cm-1间、1706~1646 cm-1间吸收峰随O-PA的变化有所不同。此外,O-PA中N-H弯曲吸收峰的出峰位置在1610~1500 cm-1之间而与PBa中苯环骨架吸收峰重叠,也使各固化体系在1617~1447 cm-1范围内的吸收峰随O-PA含量的不同而略有不同。同时,各固化体系吸收峰的位置和形状基本一致,表明O-PA的引入不改变苯并噁嗪固化物的化学结构。
图6
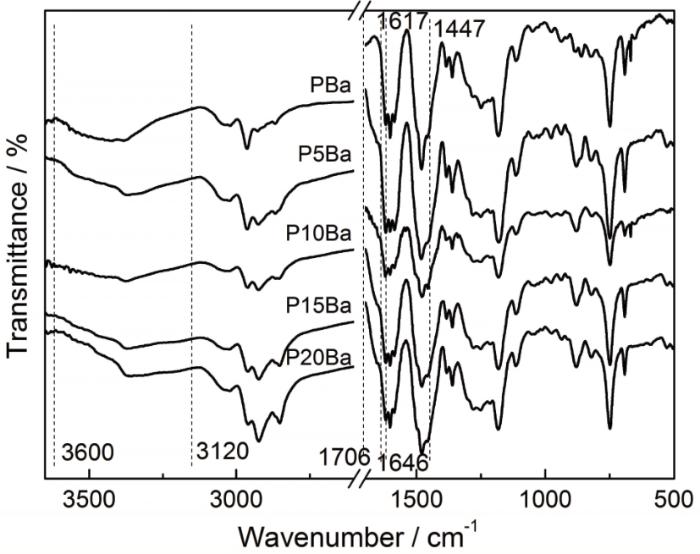
2.4 聚酰胺650对苯并噁嗪韧性的影响
聚酰胺650分子中较长的脂肪碳链具有增塑作用,可有效改善环氧树脂的韧性,因而也可能提高聚苯并噁嗪的韧性。本文进行冲击性能测试研究了O-PA对Ba和Pddm聚合物韧性的影响,结果如图7所示。可以看出,随着O-PA含量的提高PBa和PPddm的冲击强度不断提高,即O-PA对聚苯并噁嗪有良好的增韧效果。尤其是PBa,当固化体系中O-PA的含量达到20%,其冲击强度由最初的1.63 kJ/m2增加到5.75 kJ/m2。
图7
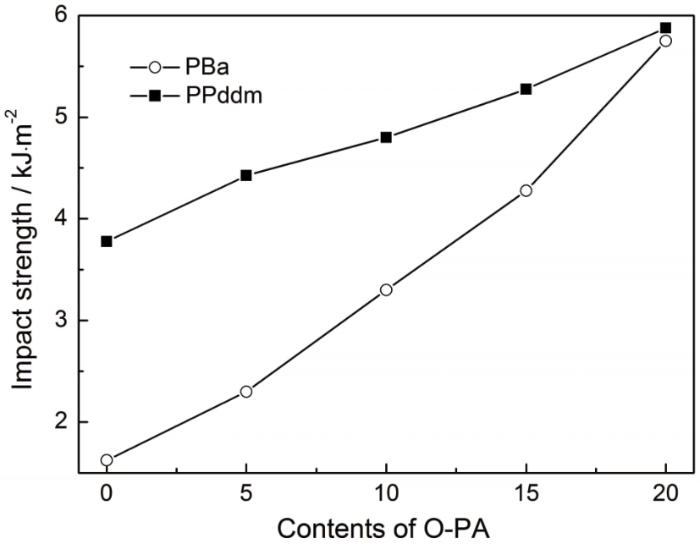
图7
O-PA对聚苯并噁嗪冲击强度的影响
Fig.7
Effect of O-PA on the impact strength of polybenzoxazine
2.5 聚酰胺对苯并噁嗪热稳定性影响
图8和表3为聚酰胺650对PBa热稳定性影响的TGA测试结果。为便于比较,O-PA的TGA结果也一并列出。这些结果表明,随着O-PA的引入PBa的初始失重温度和800℃残炭率都有所降低。DTG曲线表明,PBa及其聚酰胺共混物均表现出相似的三阶段失重模式,说明O-PA的引入没有改变PBa的化学交联结构。其中位于100~260℃间的失重主要是小分子的挥发造成,其原因可能是引入O-PA使吸附水增加(样品测试前仅在100℃干燥1 h)。水在此区间的挥发引起共混物的初始失重温度向低温处移动。发生在250~330℃间的第二阶段失重随O-PA的引入而有所增加。以往的研究结果表明,此阶段的失重主要是苯及其衍生物、胺类等的挥发,应该是O-PA与聚苯并噁嗪中Mannich桥上的N原子形成的氢键削弱了与之相连的C-N键,使小分子降解产物(包括胺类、苯类等)较易产生和挥发。但是,O-PA也使PBa在330~380℃间的失重速率降低。研究表明,在此温度段内的降解产物主要为酚类、胺类等[19],可能是O-PA与酚羟基间的氢键抑制了酚类的挥发。DTG曲线表明,O-PA的降解和挥发主要发生在350~500℃,使PBa混合体系在第三失重阶段(400~500℃)的失重速率比PBa增大,且O-PA越多则固化体系的失重速率越大。
图8
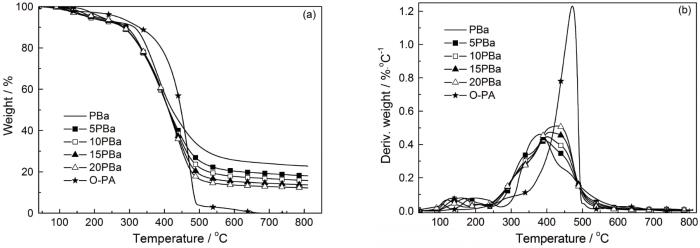
图8
O-PA对PBa热稳定性的影响
Fig.8
Effect of O-PA on the thermal stability of PBa (a) TGA curves; (b) DTG curves
表3 O-PA对聚苯并噁嗪热稳定性影响的TGA测试结果
Table 3
| Sample | PBa | 5PBa | 10PBa | 15PBa | 20PBa | PPddm | 5PPddm | 10PPddm | 15PPddm | 20Pddm | PPa | 5PPa | 10PPa | 15PPa | 20PPa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T5%/℃ | 218.6 | 194.6 | 180.1 | 188.6 | 205.5 | 300.4 | 190.7 | 228.9 | 247.9 | 255.6 | 205.0 | 165.1 | 142.3 | 206.8 | 158.0 |
| T10%/℃ | 313.8 | 292.2 | 281.3 | 281.6 | 283.9 | 358.3 | 328.5 | 327.2 | 320.1 | 310.7 | 315.9 | 302.9 | 289.7 | 314.8 | 294.9 |
| CY/% | 22.9 | 18.2 | 15.9 | 13.8 | 12.3 | 37.3 | 31.4 | 27.5 | 25.5 | 23.5 | 29.2 | 25.9 | 24.5 | 22.5 | 20.4 |
总之,尽管O-PA的引入没有引起PBa化学交联结构的改变,但是其与PBa中O、N原子间氢键的形成使PBa的热稳定性略有改变。
为了进一步说明聚酰胺650对聚苯并噁嗪热稳定性的影响,进行了PPddm、PPa及其O-PA混合体系的TGA测试,结果如图9和表3所示。由图9a可见,PPddm的DTG曲线与O-PA/PPddm的相似,即PPddm的失重模式和结构没有随O-PA的引入而改变。但是在390~490℃聚苯并噁嗪的热失重速率随O-PA的加入而明显增加,且O-PA的含量越多其热失重速率越大,这是O-PA的失重引起的。另外,在200~350℃添加O-PA也使PPddm的失重速率略有增加。其可能的原因是,O-PA与N原子间氢键的形成削弱了C-N键,使小分子降解产物较易形成和挥发,也与O-PA对PBa的热稳定性影响一致。上述原因使PPddm的初始热失重温度向低温处移动和800℃残炭率降低。
图9
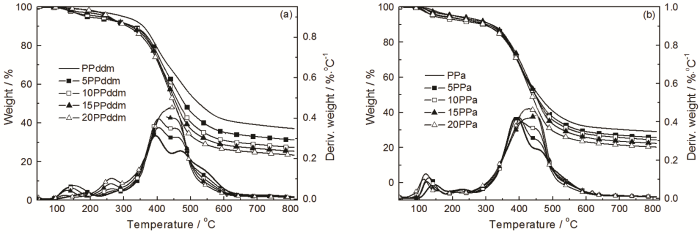
图9
O-PA/苯并噁嗪固化物的TGA测试结果
Fig.9
TGA results of the cured O-PA/benzoxazineO-PA/PPddm (a) and O-PA/PPa (b)
图9b给出了O-PA对PPa影响的TGA结果。DTG曲线表明,PPa的失重模式与O-PA/PPa混合体系相同。但是PPa在400~500℃的热失重速率增大,主要是O-PA的挥发和降解使PPa的800℃残炭率随O-PA的添加而有所降低。
上述TGA结果表明,O-PA的添加没有改变PBa、PPddm和PPa的失重模式,即聚苯并噁嗪的结构没有随O-PA的引入而改变。但是O-PA与聚苯并噁嗪中Mannich桥上N原子间氢键的形成削弱了与之相连的C-N键;加之O-PA的引入导致聚合物吸附水的增加以及高温时O-PA的降解和挥发,使聚苯并噁嗪的初始热稳定性和800℃残炭率有所降低。
3 结论
聚酰胺650与噁嗪环上O、C原子间的作用使噁嗪环较易开环,使苯并噁嗪开环聚合温度明显降低,但是O-PA的引入不影响苯并噁嗪聚合过程中的结构转变和聚合物结构。O-PA与苯并噁嗪开环聚合后酚-OH与N原子间氢键的形成抑制了苯并噁嗪链段的运动,使苯并噁嗪固化反应的活化能增大。O-PA中脂肪长链及-CONH-与苯并噁嗪间的相互作用使聚苯并噁嗪的韧性随O-PA的引入而明显提高。O-PA的引入使聚苯并噁嗪的热稳定性略有降低。表明聚酰胺650是一种性能良好的苯并噁嗪催化剂。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}