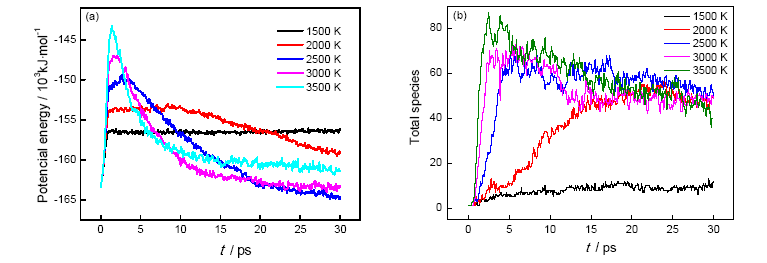
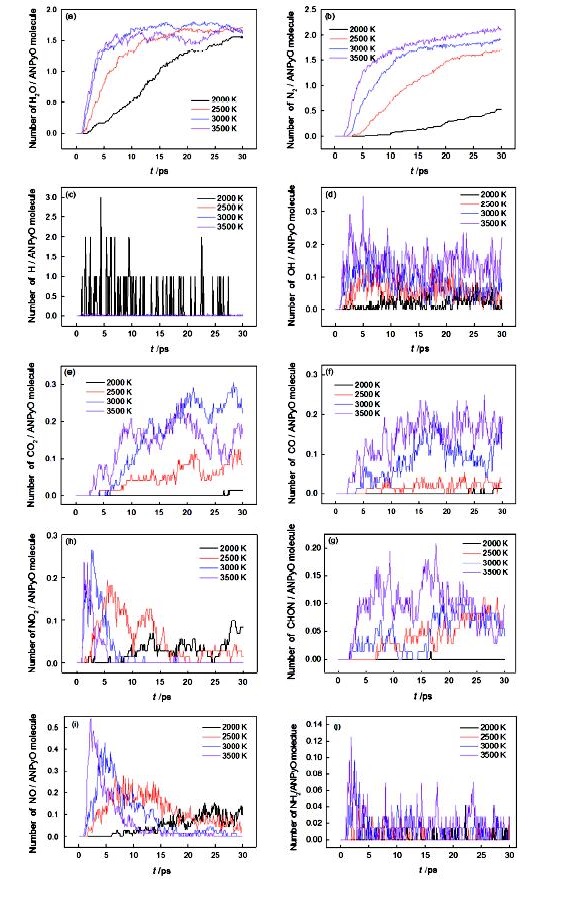
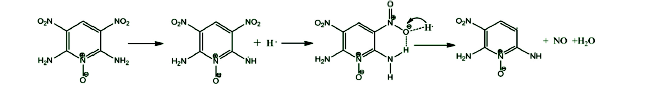
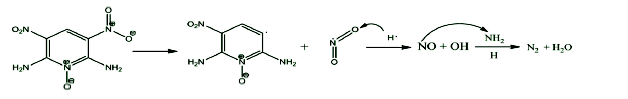
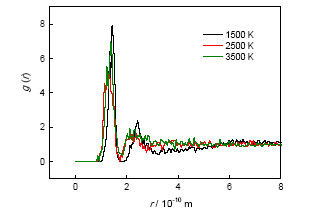
The initial decomposition of the condensed phase ANPyO crystal at various temperature (T=1500 K、2000 K、2500 K、3000 K and 3500 K) were studied by using ReaxFF reactive molecular dynamics simulation. The time evolution curve of the potential energy can be described reasonably well by a single exponential function from which the initial equilibration and induction time as well as the overall characteristic time of pyrolysis were obtained. Afterward, the activation energy Ea (88.65 kJmol-1) also was obtained from these simulations. Result show when the ANPyO molecules in the unit cell almost decomposed, the potential energy of the system significantly attenuated. Meanwhile ANPyO showed different reaction mechanisms at different temperatures. At lower temperatures (1500 K≤T≤2500 K) the hydrogen from NH2 transferred to ortho—NO2 and promote C—NO2 bond fission, while the H2O and NO molecules formed. At very high temperatures 2500 K≤T≤3500 K), the C-NO2 homolytic cleavage and C—NO2→C—ONO rearrangement hemolysis are thermo dynamically favorable pathways in the early thermal decomposition of ANPyO. According to calculations using limited time steps, the main products are H2O、N2、NO2、NO、CO2、CO、OH and HONO. Secondary products are mainly NO2、NO、OH and HONO, which has strong oxidizing property, so that the distribution has a dramatic fluctuation characteristics. It is found that H2O and N2 are the main stable products of thermal decomposition. Pyridine ring fission does not take place until most of the attached groups have interacted or disconnected, and increasing temperature accelerates fission of Pyridine ring and further decomposition to generate both CO2, CO, NO, and amount of carbon-containing clusters.
Key words:
other disciplines of
;
the materials science
;
high temperature thermal decomposition
;
ReaxFF potencial energy
;
ANPyO
;
molecular dynamics
Y. Q.Yang, S. F.Wang, Z. Y.Sun, D. D.Dlott, Propagation of shock-induced chemistry in nanoenergetic materials: The first micromete, J. Appl. Phys., 95(7), 3667(2004)
The propagation of shock-induced chemical reactions over nanometer distances is studied in energetic materials consisting of Al nanoparticles (30, 62, and 110 nm) in the polymer oxidizers nitrocellulose (NC) and Teflon. Picosecond laser flash heating vaporizes the Al particles, which react with surrounding oxidizer and generate a spherical shock wave with a rapidly dropping pressure, that decomposes the NC or Teflon out to a diameter d rxn . A methodology is developed to measure d rxn as a function of laser energy, that uses the average distance between nanoparticles d avg as a length scale and identifies the ablation threshold as occurring when the reaction spheres from multiple particles coalesce. At minimal laser fluences, d rxn is slightly larger than the diameter of the polymer sphere needed to just oxidize the nanoparticle. The excess diameter is attributed to the chemical energy of oxidation. At larger laser fluences where chemical energy is unimportant, d rxn 鈭滶 over the length scale of 50鈥1500 nm, where E is the energy in the spherical shock. Shock-induced chemical reactions propagate farther with larger nanoparticles and farther in Teflon than in NC. The linear dependence of d rxn on E is explained using a hydrodynamic model that assumes chemistry occurs when a pressureP is applied for a given time t, so that Pt= constant .
F.Castro-Marcano, A. M.Kamat, M. F.Russo, A.C.T. van Duin, J. P. Mathews Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field, J.Combustion and Flame, 159(3), 1272(2012)
Coal or biomass chars are complex carbonaceous materials that are important energy sources for electricity production. Reactive molecular dynamics simulations are a useful tool to examine the chemical reactions occurring in complex processes, providing that a realistic structural representation and an applicable reactive force field can be utilized. Combustion of coal (or biomass) char is one such area were additional insight would be helpful for utilization enhancements and pollution control. In this investigation a devolatilized Illinois No. 6 coal char atomistic representation was generated, using Fringe3D and additional Perl scripts, coupled with the ReaxFF reactive force field for hydrocarbon combustion. Fringe3D facilitates the char structure generation process by producing a distribution of aromatic structures based on HRTEM lattice fringe image analyses. Perl scripts were used for incorporating heteroatoms and aliphatic components to aid elimination of investigator bias, and facilitate a more rapid construction process. The char structure was constrained by a combination of elemental and NMR literature data. Chemical and physical parameters were found to be broadly consistent with the experimental data. The ReaxFF force field for hydrocarbon combustion was used to perform simulations to examine the structural transformations and chemical processes associated with char combustion. In this initial work, very high temperatures (3000-4000 K) were selected for ReaxFF simulation under stoichiometric, fuel lean and rich combustion conditions. These elevated temperatures were chosen to observe chemical reactions proceed to completion within a computationally practical simulation time. Analyses indicated that char oxidation process was primarily initialized by either thermal degradation of char structure to form small fragments, that were subsequently oxidized, or by hydrogen abstraction reactions by oxygen molecules and 0 and OH radicals. A more rapid oxidation and combustion of the polyaromatic structures occurred at fuel lean (oxygen rich) conditions compared with fuel rich combustion. Char transitions included 6-membered ring conversion into 5- and 7-membered rings that further decomposed or reacted with mostly O and OH radicals. This work further demonstrates the utility of ReaxFF force field integration with representative char structural models to investigate physical and chemical transformations of char structure during combustion at high-temperature conditions. (C) 2011 The Combustion Institute. Published by Elsevier Inc. All rights reserved.
L. C.Liu, C.Bai, H.Sun, W. A.Goddard, Mechanism and kinetics for the initial steps of pyrolysis and combustion of 1, 6-Dicyclopropane-2, 4-hexyne from ReaxFF reactive dynamics, J. Phys. Chem.A, 115(19), 4941(2011)
We report the kinetic analysis and mechanism for the initial steps of pyrolysis and combustion of a new fuel material, 1,6-dicyclopropane-2,4-hexyne, that has enormous heats of pyrolysis and combustion, making it a potential high-energy fuel or fuel additive. These studies employ the ReaxFF force field for reactive dynamics (RD) simulations of both pyrolysis and combustion processes for both unimolecular and multimolecular systems. We find that both pyrolysis and combustion initiate from unimolecular reactions, with entropy-driven reactions being most important in both processes. Pyrolysis initiates with extrusion of an ethylene molecule from the fuel molecule and is followed quickly by isomerization of the fuel molecule, which induces additional radicals that accelerate the pyrolysis process. In the combustion process, we find three distinct mechanisms for the O(2) attack on the fuel molecule: (1) attack on the cyclopropane, ring expanding to form the cyclic peroxide which then decomposes; (2) attack onto the central single bond of the diyne which then fissions to form two C(5)H(5)O radicals; (3) attack on the alkyne-cyclopropane moiety to form a seven-membered ring peroxide which then decomposes. Each of these unimolecular combustion processes releases energy that induces additional radicals to accelerate the combustion process. Here oxygen has major effects both as the radical acceptor and as the radical producer. We extract both the effective activation energy and the effective pre-exponential factor by kinetic analysis of pyrolysis and combustion from these ReaxFF simulations. The low value of the derived effective activation energy (26.18 kcal/mol for pyrolysis and 16.40 kcal/mol for combustion) reveals the high activity of this fuel molecule.
X. F.Ma, W. H.Zhu, J. J.Mao, H. M.Mao, Molecular dynamics study of the structure and performance of simple and double bases propellants, J. Hazard. Mater., 156(1-3), 201(2008)
To investigate the structure and performance of simple and double bases propellants, the nitrocellulose (NC), nitroglycerin (NG), and double mixed system (NC+NG) have been simulated by using the molecular dynamics (MD) method with the COMPASS force field. The interactions between NC and NG have been analyzed by means of pair correlation functions. The mechanical properties of the three model systems, i.e. elastic coefficients, modulus, Cauchy pressure, and Poisson's ratio, etc., have been obtained. It is found that the rigidity, ductibility, and tenacity of the double bases propellants (NC+NG) are stronger than those of simple base propellants (NC), which attributes to the effect of NG and the strong interactions between NC and NG. The detonation properties of the three systems have also been calculated and the results show that compared with the simple base propellant (NC), the detonation heat and detonation velocity of the double base propellants (NC+NG) are increased.
H.Ritter, H. H.Licht, Synthesis and reactions of dinitrated amino and diaminopyridines, J. Heterocycl. Chem., 32(2), 585(1995)
Abstract Nitration of amino- and diaminopyridines and -picolines led, in unexpected one-step reactions, to dinitrated derivatives. Dinitropicolines gave styrylpyridines, and 2-amino-6-hydroxy-3,5-dinitropyridine was transformed by the thermolysis of its azido derivative into 5-amino-6-nitro[1,2,5]oxadiazolo[3,4- b ]pyridine. Using 1 H and 13 C nmr spectroscopy, azido-tetrazole tautomerism of 2-amino-6-azido-3,5-dinitropyridine and intramolecular hydrogen bonding at 20掳 for several 2-amino-3,5-dinitro-6- R -pyridines have been proved.
ABSTRACT In this paper, the discovery and synthesis of the explosive 1,1-diamino-2,2-dinitroethene (FOX-7) are described, together with an account of its structural, spectroscopic, and explosive properties. The chemical reactivity of FOX-7 towards nucleophilic substitution (transamination), electrophilic substitution, and acid-base properties is explored, as is its thermal behavior (phase transformations and thermal decomposition). The molecular structure and physical properties of FOX-7 are compared with those of its three isomers (as yet unsynthesized), as derived by theoretical calculations. Finally, the physical properties of FOX-7 are compared to those of various energetic molecules that are structurally related to FOX-7.
MACongming, LIUZuliang, XUXiaojuan, YAOQizheng, Research progress on the synthesis of energetic pyridines,Chinese Journal of Organic Chemistry, 34, 1288(2014)
LIUHuaning, ZHENGYu, QIUCongli, WANGXiaoming, LIWenbin, CHENGBo, Experimental study on jet impact sensitivity of a new explosive 2, 6-diamino-3, 5-dinitropyridine-1-oxide,Chinese Jornal of Energetic Materials, 22(3), 337(2014)
Z. W.He, S. Q.Zhou, X. H.Ju, Z. L.Liu, Computational investigation on 2, 6-diamino-3, 5-dinitropyridine-1-oxide crystal, Struct. Chem., 21(3), 651(2010)
Density functional theory calculations were performed on crystalline 2,6-diamino-3,5-dinitropyridine-1-oxide (ANPyO). The conduct bands are generally quite flat, while the valence bands are uneven. The carbon, oxygen and amino nitrogen atoms make up the narrow lower energy levels. While the carbon, amino nitrogen and atoms in nitro group make up the higher energy levels. Change of electronic charges for the decrease of the cell edge and are almost the same, but different from the decrease of the cell edge , indicating an anisotropic effect related to compressions. The C-Nitro and the N-O (-oxide) bonds are the weakest, and tend to rupture upon external stimulation. The Mulliken population for the N-O (-oxide) bond in crystal is much smaller than that in molecule, indicating that the molecular packing weakens this bond. Judged by the fact of N-O (-oxide) bond being weaker than C-Nitro bond, ANPyO is sensitive to mechanic impact than 1,3,5-triamino-2,4,6-trinitrobenzene, which is in good agreement with experiment. The crystal lattice energy is predicted to be 鈭166.03 kJ/mol, after being corrected for basis set superposition error.
A. C.T.Van Duin, S. Dasgupta, F. Lorant, W. A. Goddard, ReaxFF: A reactive force field for hydrocarbons, J. Phys. Chem.A, 105(41), 9396(2001)
[本文引用:1]
13
G.Chevrot, A.Sollier, N.Pineau, Molecular dynamics and kinetic study of carbon coagulation in the release wave of detonation products, J. Chem. Phys., 136(8), 191(2012)
ABSTRACT We present a combined molecular dynamics and kinetic study of a carbon cluster aggregation process in thermodynamic conditions relevant for the detonation products of oxygen deficient explosives. Molecular dynamics simulations with the LCBOPII potential under gigapascal pressure and high temperatures indicate that (i) the cluster motion in the detonation gas is compatible with Brownian diffusion and (ii) the coalescence probability is 100% for two clusters entering the interaction cutoff distance. We used these results for a subsequent kinetic study with the Smoluchowski model, with realistic models applied for the physical parameters such as viscosity and cluster size. We found that purely aggregational kinetics yield too fast clustering, with moderate influence of the model parameters. In agreement with previous studies, the introduction of surface reactivity through a simple kinetic model is necessary to approach the clustering time scales suggested by experiments (1000 atoms after 100 ns, 10 000 atoms after 1 mu s). However, these models fail to reach all experimental criteria simultaneously and more complex modelling of the surface process seems desirable to go beyond these current limitations.
S.Agrawalla, A. C.T.Van Duin, Development and application of a ReaxFF reactive force field for hydrogen combustion, J. Phys. Chem.A, 115(6), 960(2011)
To investigate the reaction kinetics of hydrogen combustion at high-pressure and high-temperature conditions, we constructed a ReaxFF training set to include reaction energies and transition states relevant to hydrogen combustion and optimized the ReaxFF force field parameters against training data obtained from quantum mechanical calculations and experimental values. The optimized ReaxFF potential functions were used to run NVT MD (i.e., molecular dynamics simulation with fixed number of atoms, volume, and temperature) simulations for various H(2)/mixtures. We observed that the (HO(2)) radical plays a key role in the reaction kinetics at our input conditions (T 鈮 3000 K, P > 400 atm). The reaction mechanism observed is in good agreement with predictions of existing continuum-scale kinetic models for hydrogen combustion, and a transition of reaction mechanism is observed as we move from high pressure, low temperature to low pressure, high temperature. Since ReaxFF derives its parameters from quantum mechanical data and can simulate reaction pathways without any preconditioning, we believe that atomistic simulations through ReaxFF could be a useful tool in enhancing existing continuum-scale kinetic models for prediction of hydrogen combustion kinetics at high-pressure and high-temperature conditions, which otherwise is difficult to attain through experiments.
Q.An, S. V.Zybin, W. A.Goddard, A.Jaramillo-Botero, M.Blanco, S. N.Luo, Elucidation of the dynamics for hot-spot initiation at nonuniform interfaces of highly shocked materials, Phys. Rev.B, 84, 220101(2011)
The fundamental processes in shock-induced instabilities of materials remain obscure, particularly for detonation of energetic materials. We simulated these processes at the atomic scale on a realistic model of a polymer-bonded explosive (3,695,375 atoms/cell) and observed that a hot spot forms at the nonuniform interface, arising from shear relaxation that results in shear along the interface that leads to a large temperature increase that persists long after the shock front has passed the interface. For energetic materials this temperature increase is coupled to chemical reactions that lead to detonation. We show that decreasing the density of the binder eliminates the hot spot.
J.Budzien, A. P.Thompson, S. V.Zybin, Reactive molecular dynamics simulations of shock through a single crystal of pentaerythritol tetranitrat, J. Phys. Chem.B, 113(40), 13142(2009)
Large-scale molecular dynamics simulations and the reactive force field ReaxFF were used to study shock-induced initiation in crystalline pentaerythritol tetranitrate (PETN). In the calculations, a PETN single crystal was impacted against a wall, driving a shockwave back through the crystal in the [100] direction. Two impact speeds (4 and 3 km/s) were used to compare strong and moderate shock behavior. The primary difference between the two shock strengths is the time required to exhibit the same qualitative behaviors with the lower impact speed lagging behind the faster impact speed. For both systems, the shock velocity exhibits an initial deceleration due to onset of endothermic reactions followed by acceleration due to the onset of exothermic reactions. At long times, the shock velocity reaches a steady value. After the initial deceleration period, peaks are observed in the profiles of the density and axial stress with the strongly shocked system having sharp peaks while the weakly shocked system developed broad peaks due to the slower shock velocity acceleration. The dominant initiation reactions in both systems lead to the formation of NO(2) with lesser quantities of NO(3) and formaldehyde also produced.
A.Strachan, E. M.Kober, A. C.T.Van Duin, J. Oxgaard, W. A. Goddard, Thermal decomposition of RDX from reactive molecular dynamic, J. Chem. Phys., 122(5), 054502(2005)
We use the recently developed reactive force field ReaxFF with molecular dynamics to study thermal induced chemistry in RDX [cyclic-[CH2N(NO2)]3] at various temperatures and densities. We find that the time evolution of the potential energy can be described reasonably well with a single exponential function from which we obtain an overall characteristic time of decomposition that increases with decreasing density and shows an Arrhenius temperature dependence. These characteristic timescales are in reasonable quantitative agreement with experimental measurements in a similar energetic material, HMX [cyclic-[CH2N(NO2)]4]. Our simulations show that the equilibrium population of CO and CO2 (as well as their time evolution! depend strongly of density: at low density almost all carbon atoms form CO molecules; as the density increases larger aggregates of carbon appear leading to a C deficient gas phase and the appearance of CO2 molecules. The equilibrium populations of N2 and H2O are more insensitive with respect to density and form in the early stages of the decomposition process with similar timescales.
J.Quenneville, T. C.Germann, A. P.Thompson, Molecular dynamics studies of thermal induced chemistry in TATB,Shock Compression of Condensed Matter, 955(1), 451(2007)
Abstract A reactive force field (ReaxFF) is used with molecular dynamics to probe the chemistry induced by intense heating (‘accelerated cook‐off’) of 1,3,5‐triamino‐2,4,6‐trinitrobenzene (TATB). Large‐system simulations are desired for TATB because of the high degree of carbon clustering expected in this material. Using small, 32‐molecule simulations, we calculate the reaction rate as a function of temperature and compare the Arrhenius‐predicted activation energy with experiment. Decomposition product evolution (mainly N2, H2O, CO2 and graphitic carbon clusters) is followed using a 576‐molecule larger simulation, which also illustrates the effect of system size on both carbon clustering and reaction rate.
L.Zhang, S.V.Zybin, A.C.T.Van-Duin, S. Dasgupta, W. A. Goddard, Carbon cluster formation during thermal decomposition of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7- tetrazocine and 1, 3, 5-triamino-2, 4, 6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations, J. Phys. Chem.A, 113(40), 10619(2009)
T. van Duin, J. P. Mathews Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field, J.
Mechanism and kinetics for the initial steps of pyrolysis and combustion of 1, 6-Dicyclopropane-2, 4-hexyne from ReaxFF reactive dynamics, J. Phys. Chem.
Van-Duin, S. Dasgupta, W. A. Goddard, Carbon cluster formation during thermal decomposition of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7- tetrazocine and 1, 3, 5-triamino-2, 4, 6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations, J. Phys. Chem.


 , 王晓鸣
, 王晓鸣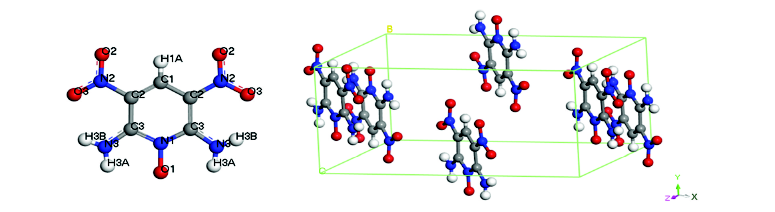



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}