
陈晓东, 辛梅华, 李明春 , 陈彰旭, 陈志强
, 陈彰旭, 陈志强
华侨大学材料科学与工程学院 环境友好功能材料教育部工程中心 厦门 361021
CHEN Xiaodong, XIN Meihua, LI Mingchun, CHEN Zhangxu, CHEN Zhiqiang
文献标识码: TB321, TQ132
文章编号: 1005-3093(2016)01-0031-07
通讯作者:
收稿日期: 2015-02-4
网络出版日期: 2016-01-25
版权声明: 2016 《材料研究学报》编辑部 《材料研究学报》编辑部
基金资助:
展开
摘要
制备了N-琥珀酰基-O-羟丙基磺酸壳聚糖(SA-HPSCS), 用FTIR、1H NMR等测试手段对其表征, 并以其为有机基质仿生合成碳酸钙, 对照比较在纯水体系和SA-HPSCS体系下合成的碳酸钙的晶型和形貌, 并考察体系的pH值对SA-HPSCS仿生合成碳酸钙的影响, 探讨其作用机理。结果表明, 在纯水体系中形成单一的方解石晶体, 而在SA-HPSCS体系中形成以球霰石为主的碳酸钙晶体。在pH=6时, SA-HPSCS体系中形成的球霰石含量达95.8%; 随着pH由6上升到10, 球霰石含量由95.8%下降到75.2%, 尺寸由6-12 μm下降到2-6 μm。这些结果说明, SA-HPSCS对碳酸钙有良好的调控作用, 形成尺寸不一、形貌差异、晶相含量可控的碳酸钙晶体。其调控作用受体系pH值的影响。
关键词:
Abstract
N-succinyl-O-hydroxypropyl sulfonated chitosan (SA-HPSCS) was prepared and then characterized by FT-IR and 1HNMR. The role of SA-HPSCS in the biomimetic synthesis of CaCO3 was studied, while the CaCO3 crystal produced in a pure water system was used as a reference. The influence of pH value on the morphology and crystallographic structure of CaCO3 crystal in the SA-HPSCS system was investigated and the relevant mechanism was proposed. The results show that the calcite is the only phase of CaCO3 crystal prepared in the pure water system, while the vaterite is dominant phase of those prepared in the SA-HPSCS system. By pH=6 the vaterite content in the prepared CaCO3 reached 95.8%, but both the size and the content of the vaterite dropped with the increase of pH value. The results also indicate that SA-HPSCS could regulate effectively the synthesis process of the CaCO3 crystal in terms of morphology, polymorph and dimension, and the pH value could strongly affect its regulation effect.
Keywords:
生物矿化是生物体利用生物大分子与矿物离子在界面处的相互作用调控矿物的成核与生长, 形成具有特定晶型、结构和理化性质的生物矿物的过程[1]。碳酸钙是自然界中储量丰富的生物矿物, 也是重要的工业原料。碳酸钙主要有3种不同的晶型, 即方解石、文石和球霰石。其中球霰石具有较高的水溶性、孔隙率, 在生物医疗和工业中有着广泛的应用[2]。但是球霰石是热力学不稳定的晶型, 易转化成热力学稳定的方解石。因此, 根据生物矿化原理以有机大分子为有机基质合成具有特定晶型的碳酸钙, 有重要的意义。
目前用于碳酸钙仿生合成的有机基质, 有多糖、蛋白质、双亲水性嵌段共聚物等[3]。作为自然界储量丰富的多糖, 壳聚糖及其衍生物在调控碳酸钙的矿化过程中扮演重要的角色。Yang等[4]以N-琥珀酰基-O-2-羟丙基-3-丁基壳聚糖为有机基质, 用气体扩散的方法制备了椭圆体和哑铃状的碳酸钙晶体。本文作者所在课题组[5]以N-组氨酸壳聚糖为有机模板调控合成碳酸钙。结果表明, N-组氨酸壳聚糖体系具有pH值响应性, 体系pH值的变化改变了N-组氨酸壳聚糖的带电性质和电荷密度, 从而影响其对碳酸钙合成的调控效果。羧基和磺酸基是生物大分子如蛋白质、多糖中常见的官能团, 能与矿物离子发生特殊的绑定作用, 从而控制矿物晶体的晶型和形貌等。本文使琥珀酸酐和3-氯-2-羟基丙磺酸钠与壳聚糖反应制备含有羧基和磺酸基的壳聚糖衍生物(SA-HPSCS)用于仿生合成碳酸钙, 研究体系pH值对SA-HPSCS仿生合成碳酸钙的影响。
壳聚糖(CS,
Bruker Avance III 型(500 MHz)核磁共振波谱仪(Bruker); Nexusu 470 型傅里叶变换红外光谱仪(Nicolet); FD-1B-50 型冷冻干燥机; PB-10 pH计; ZEN3600 纳米激光粒度及Zeta电位分析仪(Malvern); SU8010场发射扫描电子显微镜(FESEM)。
参考文献[6]制备琥珀酰化壳聚糖。将2 g壳聚糖分散于40 mL的二甲基亚砜中, 在65℃水浴下加入2.4 g丁二酸酐, 搅拌回流12 h。将过滤后得到的粉末浸泡于乙醇1 h, 过滤后用丙酮索提2 d, 70℃真空干燥, 得到琥珀酰化壳聚糖(SACS)。
将0.5 g SACS溶于30 mL 2 mol·L-1的NaOH溶液, 在70℃水浴碱化1 h后加入3.2 g 3-氯-2-羟基丙磺酸钠, 搅拌回流24 h后用去离子水透析3 d。浓缩后冷冻干燥得N-琥珀酰基-O-羟丙基磺酸壳聚糖(SA-HPSCS)。
纯水体系下碳酸钙的合成: 用去离子水配制40 mL 0.10 molL-1的CaCl2溶液, 用1 molL-1的HCl和NaOH溶液调节pH=7, 在25℃水浴中滴加40 mL 0.10 molL-1的Na2CO3溶液, 陈化4 h后过滤。将产品用去离子水和乙醇充分洗涤, 在70℃真空干燥后得到碳酸钙产品C0。
SA-HPSCS体系下碳酸钙的合成: 为考察pH对SA-HPSCS仿生合成碳酸钙晶体的影响, 在不同pH条件下(pH=6、7、8、9、10)合成5种不同的碳酸钙产品(C1、C2、C3、C4、C5)。具体合成步骤: 用去离子水配制40 mL 0.10 molL-1的CaCl2溶液, 在其中加入0.02 g的SA-HPSCS, 用1 molL-1的HCl和NaOH溶液调节pH值至特定数值, 然后在25℃水浴中滴加40 mL 0.10 molL-1的Na2CO3溶液, 陈化4 h后过滤, 再用去离子水和乙醇洗涤, 最后在70℃真空干燥, 得到碳酸钙产品。
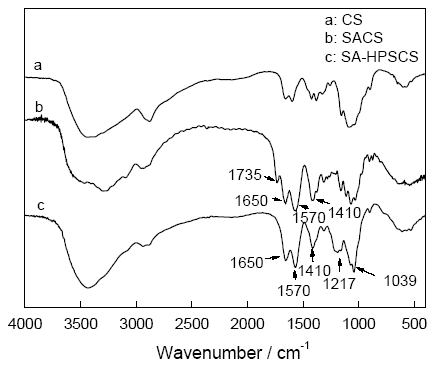
2.1.1 FT-IR分析 采用KBr压片法测得壳聚糖(CS), 琥珀酰化壳聚糖(SACS), N-琥珀酰基-O-羟丙基磺酸壳聚糖(SA-HPSCS)的红外光谱如图1所示。在图1中, 曲线b与a 相比, b在1650 cm-1和1570 cm-1处出现强的吸收峰, 分别对应酰胺Ⅰ和酰胺Ⅱ的吸收峰, 而1410 cm-1是COOH的对称振动峰[7], 1735 cm-1是酰亚胺的吸收峰, 说明琥珀酰基已引入壳聚糖分子链的氨基中。此外曲线c在1217 cm-1 和1039 cm-1处出现了较强的吸收峰, 分别对应磺酸基中O=S=O的不对称和对称伸缩振动的特征吸收峰[8]。说明磺酸基已引入, 产物为目标产物。
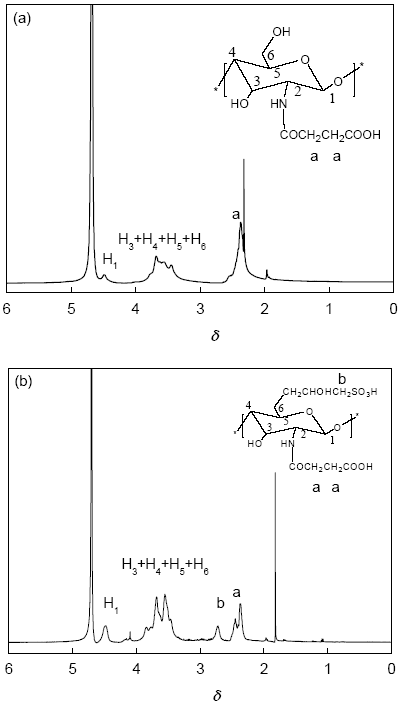
2.1.2 1H NMR分析 将琥珀酰化壳聚糖(SACS)和N-琥珀酰基-O-羟丙基磺酸壳聚糖(SA-HPSCS)分别溶于D2O中测定其1H NMR, 如图2所示。图2a给出了SACS的1H NMR图, δ=4.5是壳聚糖H1的质子峰, δ=3.70-3.88处为壳聚糖上H3、H4、H5、H6处的质子峰[9], δ=2.39是琥珀酰基中-CH2的质子峰[7], δ=2.33则为琥珀酰亚胺中的亚甲基的质子峰。图2b给出了SA-HPSCS的1H NMR图, δ=2.78为羟丙基磺酸基中与硫原子相连的—CH2的质子峰[10]。由1H NMR进一步说明壳聚糖衍生物SA-HPSCS已成功合成。在图2b中, 通过琥珀酰基中亚甲基的质子峰(δ=2.28-2.53)和羟丙基磺酸基中与硫原子相连的亚甲基的质子峰(δ=2.65-2.80)的积分面积与壳聚糖H1峰(δ=24.39-4.60)的积分面积相比, 计算出SA-HPSCS中琥珀酰基和羟丙基磺酸基的取代度分别为0.48和0.35。
图2 壳聚糖衍生物SACS和SA-HPSCS的1H NMR图
Fig.2 1H NMR spectra of chitosan derivate (a) SACS, (b) SA-HPSCS
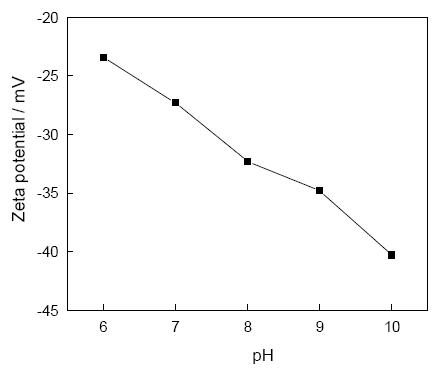
2.1.3 Zeta电位分析 N-琥珀酰基-O-羟丙基磺酸壳聚糖的Zeta电位分析结果, 如图3所示。由图可见, SA-HPSCS的Zeta电位随pH值的上升而降低, pH=6、 7、 8、 9、 10时其Zeta电位分别为-23.4、 -27.3、 -32.3、 -34.8和-40.3 mV。其原因是, SA-HPSCS的羧基和磺酸基属于阴离子基团, 碱性条件有利于基团的离子化而带负电荷[10]。因此, 随着体系的pH上升, SA-HPSCS的Zeta电位更负。这表明, 调整体系pH值可控制SA-HPSCS的带电环境, 而SA-HPSCS带电环境的变化影响其在碳酸钙矿化过程中与钙离子的静电作用, 从而影响其对碳酸钙合成的调控作用。
图3 SA-HPSCS的Zeta电位与pH值的关系
Fig.3 pH value-dependent Zeta potential curve of SA-HPSCS
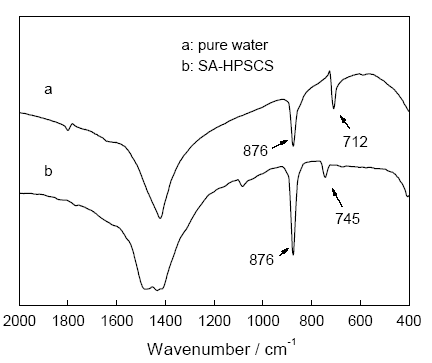
2.2.1 不同体系中碳酸钙的仿生合成 图4给出了不同体系中得到的碳酸钙的FTIR图, 曲线a, b分别对应纯水(pure water)体系和SA-HPSCS体系下得到的碳酸钙产品C0和C2。曲线a在876和712 cm-1的强烈吸收峰为碳酸钙方解石的特征峰, 曲线b除了在876 cm-1 的吸收峰外, 在745 cm-1 还出现了球霰石的特征峰[11]。这说明, 用纯水体系得到的碳酸钙为方解石, 而在体系中加入SA-HPSCS后碳酸钙主要为球霰石晶型。
图4 在不同体系中制备的碳酸钙的FTIR谱
Fig.4 FT-IR spectra of calcium carbonate obtained in different system
进一步用XRD分析上述两种体系下得到的碳酸钙产品, 结果如图5所示。图5a和b分别给出了产品C0和C2的XRD谱。图5a中主要衍射峰位2θ=23.0o、29.4o、31.4o、35.9o、39.4o、43.1o、47.5o、48.5o与标准PDF卡片(卡号为05-0586)对照, 分别对应方解石(012)、(104)、(006)、(110)、(113)、(202)、(018)、(116)晶面。在SA-HPSCS体系中制备的碳酸钙产品的XRD谱(图5b)中, 除了方解石的特征峰2θ=23.0o、29.4o、31.4o、35.9o、39.4o外, 还出现了2θ=24.9o、27.0o、32.8o的特征峰。对照标准PDF卡片(卡号为33-0268)可知, 它们分别对应球霰石(110)、(112)、(114)晶面。这表明, 在纯水体系中只能得到单一的方解石; 加入SA-HPSCS后得到的碳酸钙晶型发生变化, 其中2θ为29.4o对应的方解石(104)晶面的峰强度明显降低, 而2θ为24.9o对应的球霰石(110)晶面的峰强度明显增强。
图5 在纯水和SA-HPSCS体系中制备的碳酸钙的XRD谱
Fig.5 XRD spectra of calcium carbonate obtained in pure water (a) and SA-HPSCS (b)
可使用方程式[12]:
计算两种晶型的相对含量。方程式(1)可转换成
式中Xv/Xc表示球霰石和方解石的相对含量, Ic104和Iv110代表方解石(104)晶面和球霰石(110)晶面的衍射峰强度, Q是球霰石在混合晶型中所占的质量分数或物质的量分数。
使用式(2)计算出碳酸钙产品C2的球霰石含量为94.3%, C0的球霰石含量为0%, SA-HPSCS体系下得到的碳酸钙以球霰石为主, 与C0和C2的FT-IR分析结果一致。
图6给出了产品C0和C2的FESEM图。图6a中碳酸钙呈现出层状堆叠的立方体形状, 为方解石的特征形貌; 而图6b中碳酸钙的形貌以球体为主, 为球霰石的特征形貌。这与公式(2)的计算结果相一致。
图6 在纯水和SA-HPSCS体系中制备的碳酸钙的FESEM图
Fig.6 FESEM images of calcium carbonate in pure water (a) and SA-HPSCS (b)
综上所述, 在纯水体系中合成的是立方体形貌的方解石, 而在SA-HPSCS作用下碳酸钙则以球状的球霰石为主, 证明SA-HPSCS能有效调控碳酸钙晶型和形貌变化。
2.2.2 pH对SA-HPSCS仿生合成碳酸钙的影响 图7给出了SA-HPSCS投加量为0.02 g, 陈化时间为4 h, 在不同pH值条件下制备的碳酸钙产品的XRD谱。由图7中的衍射峰位可见, 在不同pH条件下得到的碳酸钙产品均为方解石和球霰石的混合物, 碳酸钙晶面的衍射峰强度随体系pH的变化而变化; 随着体系中pH增大, 方解石(C104)的衍射峰也随之增强。
由公式(2)计算出C1、C2、C3、C4、C5中球霰石的含量分别为95.8%、94.3%、92.1%、86.8%、75.2%。这个结果表明, 随着pH上升, 由SA-HPSCS调控得到的碳酸钙产品中球霰石的含量下降。Song等[11]使用含羧基的共聚物在不同pH条件下仿生合成碳酸钙, 也得到相似结果。他们认为, 在碱性条件下碳酸钙成核速率过高导致部分碳酸钙晶体前驱物在与有机基质发生作用前便成核生长转化为方解石, 导致球霰石的含量降低。
由图8可见, 在不同pH条件下制备的碳酸钙产品有球形和立方形。这表明, 合成的碳酸钙晶型为方解石和球霰石, 与上述相应产品的XRD分析结果一致。此外, 从图8还可以看出, 产品C1、C2、C3、C4、C5的球霰石球径不同, C1、C2、C3的球霰石球径分布不均匀, 在6-12 μm左右。当pH上升到9时, C4的球霰石球径下降到5-11 μm; 当pH=10时C5的球霰石球径下降到2-6 μm。这些结果表明, 在强碱性条件下SA-HPSCS对碳酸钙的生长具有较强抑制作用。在碱性条件下羧基等负离子基团去离子作用的加深、带电强度加强, 使其能更有效的吸附于球霰石晶体, 从而加强了对晶体的生长抑制作用[4]。因此, SA-HPSCS体系的pH越大制备出的球霰石晶体越小。
图8 pH值不同时碳酸钙的FESEM图
Fig.8 FESEM images of calcium carbonate at various pH, (a) pH 6, (b) pH 7, (c) pH 8, (d) pH 9, (e) pH 10
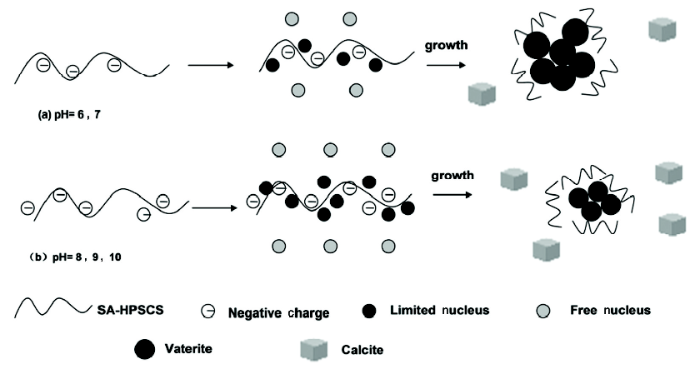
根据Ostwald step rule[13], 在过饱和溶液中先形成无定型的碳酸钙沉淀, 进而转变为亚稳态的球霰石, 最终转变为热力学稳定的方解石。在SA-HPSCS体系中, 当体系处于中性或弱酸性时(pH=7或6), SA-HPSC 的Zeta电位为负(见2.1.3), SA-HPSCS分子链上的羧基和磺酸基基团会吸引Ca2+, 致使SA-HPSCS表面局部钙离子浓度升高。此时加入CO32-与SA-HPSCS表面的钙离子作用, 形成较为稳定的晶核, 称为束缚晶核。部分钙离子脱离SA-HPSCS的束缚与CO32-形成能量较高的晶核, 称为自由晶核。束缚晶核和自由晶核, 均生长成亚稳态的球霰石。为了降低表面能, 不受SA-HPSCS作用的球霰石会在很短的时间内生长并转变成热稳态的方解石; 而对于表面吸附SA-HPSCS的球霰石, 有机基质的吸附降低了球霰石的表面能[1], 使球霰石晶体的生长和晶型转变迟滞。因此, 在SA-HPSCS调控作用下形成的球霰石会在一定时间内保持晶体的尺寸和晶型(图9a)。
图9 不同pH值时SA-HPSCS仿生合成球霰石机理
Fig.9 Schematic diagram of mechanism for synthesis of vaterite by SA-HPSCS at various pH
当体系的pH上升(pH=8-10)后, 一方面, SA-HPSCS的Zeta电位向负电位方向发展, 带电强度加强, 提高了SA-HPSCS对碳酸钙晶体的吸附能力。此时SA-HPSCS对所吸附的球霰石的抑制作用增强, 得到尺寸更小的球霰石球体; 另一方面, 体系pH上升提高了碳酸钙的过饱和浓度[11]。过饱和度S的变化与晶核生成的数目N和生成速率dr/dt相关[14]:
式中K、P、Q、β、
SA-HPSCS的羧基和磺酸基通过与溶液中钙离子作用调控碳酸钙晶核的生长和晶型的转变, 使体系形成以球霰石为主的碳酸钙晶体。体系pH 值的上升提高了SA-HPSCS的带电强度和碳酸钙的过饱和浓度, 带电强度上升加强了SA-HPSCS对所吸附的球霰石的抑制作用, 得到尺寸更小的球体; 而碳酸钙过饱和浓度的提高使晶核数目和生成速率上升, 削弱了SA-HPSCS的调控作用, 导致体系中球霰石的相对含量下降。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
McShane, Poly(vinylsulfonic acid) assisted synthesis of aqueous solution stable vaterite calcium carbonate nanoparticles, |
| [3] |
CaCO3 crystallization and morphology control by using purified soluble protein related to shell regeneration, |
| [4] |
CaCO3 crystallization controlled by (2-hydroxypropyl-3-butoxy) propylsuccunyl chitosan, |
| [5] |
Biomimetic synthesis of coexistence of vaterite-calcite phases controlled by histidine-grafted-chitosan, |
| [6] |
Immobilization of alliinase with a water soluble-insoluble reversible N-succinyl-chitosan for allicin production,
|
| [7] |
Selevtive carboxypropinonylation of chitosan: synthesis, characterization, blood compatibility, and degradation, |
| [8] |
Sulfonation and anticoagulant activity of fungal exocellular β-(1→6)-D-glucan(lasiodiplodan), |
| [9] |
Determination of the degree of N-acetylation for chitin and chitosan by various NMR spectroscopy techniques: A review, |
| [10] |
Polymer Micelle Fornation Without Gibbs Monolayer Formation: Synthesis and Characteristic Behavior of Amphiphilic Diblock Copolymer Having Strong Acid Group, |
| [11] |
The effect of a novel polyolefine based amphiphilic copolymer on the mineralization of calcium carbonate, |
| [12] |
Calcium carbonate phase analysis using XRD and FT-Raman spectroscopy, |
| [13] |
Homogeneous nucleation and the Ostwald step rule , |
| [14] |
CaCO3 nanoparticles in N, O-carboxymethyl chitosan solution, N, O-羧甲基壳聚糖体系中纳米碳酸钙的仿生合成, |

/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}