
程正载 , 张卫星
, 张卫星
CHENG Zhengzai, ZHANG Weixing
中图分类号: TQ 426
通讯作者:
收稿日期: 2014-11-1
修回日期: 2015-01-30
网络出版日期: --
版权声明: 2015 《材料研究学报》编辑部 版权所有 2014, 材料研究学报编辑部。使用时,请务必标明出处。
基金资助:
展开
摘要
以价廉的水杨醛和TiCl4为主要原料, 合成了四种水杨醛亚胺配体(5-8)及含(双苯氧基亚胺)配体的钛配合物[O-C6H4-ortho-CH=N-2, 6-(i-Pr)2-C6H3]2TiCl2(13), [O-(5-NO2)-C6H3-ortho-CH=N-2, 6-(i-Pr)2-C6 H3]2 TiCl2(14), [O-(3, 5-di-Br)-C6H2-ortho-CH=N-2, 6-(i-Pr)2-C6H3]2TiCl2(15)和[O-(3, 5-di-C(CH3)3)-C6H2-ortho-CH= N-2, 6-(i-Pr)2-C6H3]2TiCl2 (16)。用MS、1H- NMR和元素分析等手段表征了配体5-8分子结构, 用1H-NMR和元素分析表征了配合物13-16的分子结构。经MAO (甲基铝氧烷) 活化后, 钛配合物 (13-16) 在甲苯溶剂中能有效催化乙烯聚合。在60℃, 2.0 MPa及助催化剂与主催化剂摩尔比n(MAO)/n(cat)=1500∶1条件下钛配合物(14-16)催化乙烯聚合活性为1022.73-1302.27 gPE/(mmolTihMPa)), 远高于无取代水杨醛亚胺钛配合物(13)。所得聚乙烯粘均分子量(粘度法)为19266-44754, GPC测定分子量分布Mw /Mn为1.88-2.12。具有最高活性的催化剂15催化乙烯聚合得到的聚合物样品, 经13C-NMR和DSC的表征, 结果表明为线型结晶聚乙烯。
关键词:
Abstract
Four salicylaldimine ligands (called after 5, 6, 7 and 8 respectively) and their titanium complexes containing bis(phenoxy-imine) ligands, namely [O-C6H4-ortho-CH=N-2, 6-(i-Pr)2-C6H3]2TiCl2 (called after 13), [O-(5-NO2)-C6H3-ortho-CH= N-2, 6-(i-Pr)2-C6H3]2TiCl2 (called after 14), [O-(3, 5-di-Br)-C6H2-ortho-CH=N-2, 6-(i-Pr)2-C6H3]2TiCl2 (called after 15) and [O-(3, 5-di-C(CH3)3)- C6H2-ortho-CH=N-2, 6-(i-Pr)2-C6H3]2TiCl2 (called after 16) have been synthesized with salicylaldehyde and TiCl4 as raw materials. The prepared ligands and complexes were characterized by means of 1H-NMR and elemental analyses as well as mass spectra. After activated with methyaluminoxane (MAO), the complexes 13-16 become efficient catalysts for ethylene polymerization in methylbenzene. Under the conditions of T= 60℃, P=2.0 MPa and n(MAO)/n(cat)=1500∶1, the catalytic activities for the activated complexes 14-16 reached 1022.73-1302.27 gPE / (mmolTihMPa), which were much higher than that for the activated complex 13. The prepared polyethylene possessed a viscosity-average molecular weight in a rang of 19266-44754measured by viscometry and a molecular weight distribution Mw /Mn in a rang of 1.88-2.12 measured by Gel Permeation in Chromatography. Among others the activated complex 15 displays the highest activity for ethylene polymerization under the same conditions. The characterization by 13C-NMR and DSC showed that the polymer synthesized with catalyst of the activated complex 15 was actually a kind of linear and crystalline polyethylene.
Keywords:
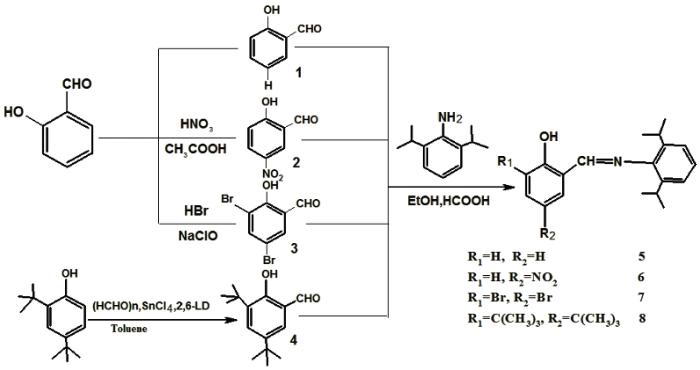
近年来, 使用苯氧基亚胺配体合成高性能聚烯烃催化剂成为烯烃配位聚合的研究热点[1]。引入具有一定空间位阻或电子效应的取代基团对配体进行结构修饰, 能裁剪聚烯烃催化剂分子结构, 制备出具有独特结构和性能的高分子材料[2]。Liu等[3] 研究了含有这类配体的钛催化剂, 将其与ZnEt2共同作用催化合成了分子量可控的双峰聚乙烯, 而且表现出较高的活性。Matoishi等[4] 考察了16种此类催化剂催化乙烯聚合的活性, 并催化合成了新的PE/PEG材料, 表现出较好的活性。Damavandi等[5]改变水杨醛亚胺基团上的配体结构合成两种新的锆基FI型催化剂, 发现将亚胺氮原子上的配体由单芳环替换为芳族稠环增加了催化剂的空间位阻, 削弱了催化活性, 但是所得聚乙烯的分子量有所增加。许多含苯氧基亚胺配体的后过渡非茂金属配合物对乙烯配位催化聚合具有与茂金属铬、锆和钛等前过渡金属配合物[6, 7]相同数量级的高活性。Younkin等[8]、Connor等[9]及Bansleben等[10]对含这类配体镍化合物催化乙烯的研究表明: 在水杨醛基上氧邻、对位引入大位阻取代基, 能使乙烯聚合活性大大提高, 而Suzuki等[11]在配体中引入强吸电子基团提高了IVB族配合物催化乙烯的聚合性能, 并最先采用水杨醛亚胺前过渡金属配合物(被称为FI催化剂)催化乙烯聚合, 发现有比茂金属化合物Cp2TiCl2和Cp2ZrCl2更高的催化聚合活性。尽管Fujita小组合成的很多锆系FI催化剂具有很高活性, 但是普通钛系FI催化剂的活性并不高[12]。在苯氧基亚胺配体上引入强吸电子的氟基F(s)后, 这类钛系FI催化剂在温和条件下表现出极高的乙烯及丙烯催化聚合活性[13, 14]。但是这类含氟FI型钛系催化剂的配体并不易得到, 致使催化剂制备成本较高, 难以实现工业化。金国新等[15]合成了含烯丙基席夫碱钛系金属配合物, 在助催化剂(MMAO)存在下, 配合物催化乙烯聚合也显示出很高的催化活性, 而合成的二双齿钛系烯烃聚合催化剂[C3H6(N=CH-Ar-O)2]TiCl2[16]除了具有高的催化活性外, 还有聚合条件温和、可以得到不同分子量的聚乙烯等特点。为了获得有较高工业化前景的高效FI催化剂, 本文以价廉易得的水杨醛为起始原料, 经过溴代、硝化等反应高收率地制备四种取代水杨醛, 通过与2, 6-二异丙基苯胺缩合, 与金属配合物TiCl4配位, 制备FI型钛系催化剂, 用于催化乙烯聚合。
实验用试剂级原料包括: 2, 6-二异丙基苯胺, 甲基铝氧烷(MAO)为1.25 mol/L甲苯溶液, 四氢呋喃(THF)、甲苯和石油醚等溶剂须先经NaOH(s)干燥数天, 后经二苯甲酮和钠回流至紫色, 在氩气保护下蒸出备用。
(1) 5-硝基水杨醛的合成
参考文献[17]中的方法合成5-硝基水杨醛, 所得目的产物为淡黄色晶体, 产率56.00%, 熔点125-127℃(文献值125-126℃), 色质联用检测其纯度为97.60%, 其余为3-硝基水杨醛。质谱主要碎片, m/z: 167 (M+, 100), 151, 137, 120, 109, 75, 65, 53, 39。
(2) 3, 5-二溴水杨醛的合成
参考文献[18] 中的方法合成3, 5-二溴水杨醛, 所得目的产物为淡黄色针状晶体, 产率83.00%, 熔点85-86℃(文献值84-86℃), 色质联用检测其纯度为98.71%。质谱主要碎片, m/z: 280 (M, 100%), 262, 252, 234, 223, 170, 155, 143, 119, 91, 74, 63, 53。
(3) 3, 5-二叔丁基水杨醛的合成
参照文献[19]中的方法合成3, 5-二叔丁基水杨醛。所得目的产物为淡黄色晶体, 产率为76.60%, 熔点58-59℃(文献值58-60℃), 质谱主要碎片, m/z: 234 (M+, 28%), 219 (M+-CH3, 100% ), 191, 163, 135, 57。1H-NMR (400MHz, CDCl3) 分析结果为: δ=11.65 (s, 1H, OH), 9.86 (s, 1H, CHO), 7.59 (d, JHH= 1H, Ar-H), 7.35 (d, JHH= 1H, Ar-H), 1.43 (s, 9H, -C(CH3)3), 1.33 (s, 9H, -C(CH3)3)。
(4) 配体5、6、7和8的合成
参考文献[20]中的方法的方法合成四种水杨醛亚胺配体。以30.0 mL乙醇为溶剂, 加入上述合成的取代水杨醛50.00 mmol, 8.90 g 2, 6-二异丙基苯胺, 0.5 mL甲酸, 在室温下搅拌10 h后生成大量的黄色粉末。冷却过滤并用冷无水乙醇洗涤后在无水乙醇中重结晶, 分别得到黄色的针状晶体5-8。收率分别为91.21%、90.80%、90.92%和92.50%, 纯度分别为98.81%、98.44%、99.13%和96.97 %。
配体5的1H-NMR分析结果为: δ=1.18 (d, JHH=4.8Hz, 12H, CH3), 3.04 (m, 2H, i-Pr-CH), 6.93 (d, JHH=4.8Hz 1H, Ar-H), 7.09 (d, JHH=3.6Hz 1H, Ar-H), 7.18 (s, 3H, Ar-H), 7.35 (d, JHH=3.6Hz, 1H, Ar-H), 7.42 (d, JHH=3.6Hz, 1H, Ar-H), 8.30 (s, 1H, CH=N), 14.98 (s, 1H, OH)。元素分析(C19H23ON, 281.40), 实测值(计算值) (%): C 81.34 (81.10), H 8.36 (8.24), N 4.80 (4.98)。
配体6的主要质谱碎片为m/z=326 (M+, 83%), 311 (M+ -CH3, 75%), 269, 252, 22, 188, 174, 146, 132, 117, 91, 77。红外分析结果为IR (cm-1): 3410, 3076, 1622, 1500, 1330, 1180, 826。1H-NMR分析结果为: δ=1.32 (d, JHH=4Hz, 12H, CH3), 3.15 (m, 2H, i-Pr-CH), 7.23-7.42 (m, 3H, Ar-H), 8.39-8.49 (m, 3H, Ar-H), 8.59 (s, 1H, CH=N), 14.28 (s, 1H, OH)。元素分析(C19H22O3N2, 326.39), 实测值(计算值) (%): C 69.71 (69.92), H 6.86(6.79), N 8.52 (8.58)。
配体7的主要质谱碎片为m/z=439 (M+, 85%), 424 (M+, -CH3, 88%), 382 (100%), 367, 301, 265, 220, 204, 188, 174, 160, 146, 132, 117, 104, 91, 77, 63, 41。1H-NMR分析结果为: δ=1.29 (d, JHH=2Hz, 12H, CH3), 3.13 (m, 2H, i-Pr-CH), 7.28 (s, 3H, Ar-H), 7.51 (s, 1H, Ar-H), 7.98 (s, 1H, Ar-H), 8.45 (s, 1H, CH=N), 14.11 (s, 1H, OH)。元素分析(C19H21Br2ON, 439.19), 实测值(计算值) (%): C 52.09 (51.96), H 4.65 (4.82), N 3.13 (3.19)。
配体8的主要质谱碎片为m/z=393 (M+, 100%), 378, 362, 350, 336, 320, 203, 188, 174, 162, 146, 132, 119, 105, 91, 77, 57, 41。1H-NMR分析结果为: δ=1.23 (d, JHH=3.6Hz, 12H, i-Pr-CH3), 1.35 (s, 18H, C(CH3)3), 3.07 (m, 2H, i-Pr-CH), 7.22 (s, 3H, Ar-H), 7.49 (s, 1H, Ar-H), 7.91 (s, 1H, Ar-H), 8.53 (s, 1H, CH=N), 14.23 (s, 1H, OH)。元素分析(C27H39ON, 393.61), 实测值(计算值) (%): C 82.56 (82.39), H 9.76 (9.99), N 3.68 (3.56)。
1.2.2 配合物的合成 采用标准Schlenk 技术, 分别按图式2路线合成四种FI型钛系催化剂13-16。
(1) 配合物13的合成 先将1.48 g (5.27 mmol) 配体5溶于20.0 mL二氯甲烷, 然后将混合溶液逐滴加入到含有5.00 g (2.64 mmol) TiCl4和30.0 mL二氯甲烷的150mL schlenk瓶中, 混合溶液逐渐变为深紫色后将其在室温下搅拌反应2 h, 减压除去溶剂后将残渣经戊烷 (10.0 mL)洗涤两次, 真空干燥后得到配体9。将所得配体9溶于50.0 mL二氯甲烷后加入0.41 g (4.00 mmol) NEt3, 在室温下继续搅拌反应4 h, 减压除去溶剂后加入100.0 mL四氢呋喃, 过滤除去NH4Cl, 滤液经减压除去溶剂, 剩余固体用戊烷 (10.0 mL)洗涤两次后得到1.13 g (1.67 mmol) 橙色固体, 收率63.38%。
(2) 配合物14的合成 在经真空抽烤和Ar置换的150 mL Schlenk瓶中加入0.02 mol NaH和50.0mL THF, 加入3.26 g (10.00 mmol) 配体6, 反应3 h。过滤以除掉未反应的NaH, 即得配体钠盐10的THF溶液。在-78℃及良好搅拌条件下将其逐滴滴加到含1.70 g (5.00 mmol) TiCl4 2THF和50.0 mL THF的溶液中。滴加完毕后逐渐升至室温, 继续搅拌反应18 h。减压抽干溶剂后加入40.0 mL二氯甲烷, 搅拌15 min。过滤后将滤渣用二氯甲烷(10.0 mL×2)洗涤, 合并滤液后在减压条件下抽干溶剂, 得到红色固体。加入20.0 mL乙醚和40.0 mL石油醚, 搅拌30 min。过滤后将滤渣用石油醚(15.0 mL×2)洗涤两次, 真空抽干溶剂后得到2.29 g (2.98 mmol) 红棕色配合物14, 收率为59.60%。
(3) 配合物15的合成 在经真空抽烤和Ar置换的150 mL Schlenk瓶中加入2.31 g (5.27 mmol)配体7和20.0 mL二氯甲烷, 在常温下逐滴滴加含有0.50 g (2.64 mmol)新蒸TiCl4和30.0 mL二氯甲烷的溶液, 将所得浅红色溶液在常温下搅拌反应2 h, 加入735.0 μL NEt3 (534.00 mg, 5.27 mmol)。继续搅拌反应2 h, 减压抽去溶剂后加入80.0 mL甲苯, 搅拌30 min。将混合液过滤除掉NH4Cl, 清液浓缩至20.0 mL后置于-20℃冰箱中, 析出红色晶体, 抽干溶剂, 用石油醚(15.0 mL×2)洗涤两次。真空抽干溶剂后得到1.89 g (1.90 mmol)红色配合物15, 收率为72.11%。
(4) 配合物16的合成 在经真空抽烤和Ar置换的150 mL Schlenk瓶中加入3.92 g (10.00 mmol)配体8, 50.0 mL THF和0.02 mol NaH在室温下反应3 h, 过滤出未反应的NaH后得到配体钠盐12的THF溶液。对所得配体钠盐12进行搅拌, 在温度为-78℃的条件下逐滴加入到含有50.0 mL THF和1.70 g(5.00 mmol)TiCl42THF的混合溶液中, 缓慢升温至室温后搅拌反应18 h, 反应完毕后在减压条件下除去溶剂, 加入40.0 mL二氯甲烷, 搅拌过滤并将所得滤渣用20.0 mL二氯甲烷洗涤, 滤液经减压抽滤后得到红色固体。使所得固体与40.0 mL石油醚和20.0 mL乙醚搅拌反应15 min, 过滤后将所得滤饼经30.0 mL石油醚洗涤2次, 减压抽滤除去溶剂后得到2.49 g (2.75 mmol)配合物16, 收率为55.00%。
乙烯加压聚合反应在装有500 r/min的搅拌器、加料管和进气管等设施的100 mL不锈钢高压釜中进行, 如图3所示。由温控系统控制反应温度, 压控阀控制反应釜内乙烯压力。聚合前将反应釜烘干, 并将反应釜及相连的系统管路抽真空除氧, 再经乙烯气置换三次。在乙烯气体保护下由加料口一次性加入混合好的催化剂和MAO的甲苯溶液(总体积30.0 mL)。加料完毕后立即关闭加料阀并通入乙烯, 控制一定的反应温度和压力。反应一定时间后, 系统放空出料。用含10%盐酸的乙醇溶液终止反应, 所得聚合物经过滤后用乙醇和纯水多次洗涤。在60℃真空干燥至恒重, 计算催化剂的活性。
用粘度法和凝胶色谱法(GPC) 测定聚乙烯分子量。前者以十氢萘为溶剂, 在(135±0.1)℃使用乌式粘度计测定[η], 按公式
计算聚乙烯的粘均分子量Mη; 后者于135℃以1, 2, 4-三氯苯为流动相在Waters-150-C- ALC/GPC仪上测定。用Perkin Elmer DSC-7差示量热扫描仪测定聚合物熔点, 扫描速率10℃/min, 扫描范围50-160℃。
在Perkin-Elmer240型元素分析仪上进行元素分析, 定量氧燃烧, 热导池检测器。用VERTEX 70型FT-IR红外光谱仪测定(KBr压片)配体和配合物的红外光谱; 用Bruker Avance DMX500型核磁共振仪检测核磁共振谱, TMS为内标, CDCl3为溶剂。在HP6890型气相色谱仪上进行气相色谱分析, 使用SE-30毛细管柱(50 m×0.2×0.5 μm), FID检测器。用HP6890/5973型色谱-质谱联用仪进行质谱测定, 电子撞击法, 电子束70 eV, 升温范围50-300℃。
采用四种不同方法将配体5-8与金属钛氯化物配位。配体5和7均与TiCl4配位形成中间络合物9和11, 再与NEt3反应形成最终配合物13和15; 而配体6和 8与NaH形成钠盐10和12, 再与TiCl4·2THF配位, 可形成最终配合物14和16。
配合物13的1H-NMR分析结果为: δ=1.13 (d, JHH=2.8 Hz, 24H, i-Pr-CH3), 3.55-3.67 (m, 4H, i-Pr-CH), 6.76 (d, JHH=4.8 Hz, 2H, Ar-H), 7.13 (d, JHH=2.8 Hz, 2H, Ar-H), 7.21 (d, JHH=3.6 Hz, 2H, Ar-H), 7.25 (d, JHH=3.6 Hz, 2H, Ar-H), 7.31 (d, JHH=4.4 Hz, 2H, Ar-H), 7.61 (d, JHH=3.2 Hz, 2H, Ar-H), 7.67 (d, JHH=4.4 Hz, 2H, Ar-H), 8.43 (m, 2H, CH=N)。元素分析(C38H44O2N2TiCl2, 679.56), 实测值(计算值) (%): C 67.42 (67.16), H 6.41 (6.53), N 4.32 (4.12)。
配合物14的1H-NMR分析结果, 如图4所示。从图4可知: δ=1.15 (d, JHH=4 Hz, 24H, i-Pr-CH3), 2.92-2.95 (m, 2H, i-Pr-CH), 3.15 (m, 2H, i-Pr-CH), 6.81-7.18 (m, 12H, Ar-H), 8.35-8.37 (m, 2H, CH=N)。元素分析(C38H42O6N4TiCl2, 769.56), 实测值(计算值)(%): C 59.64 (59.31), H 5.36 (5.50), N 7.42 (7.28)。配合物14的IR分析结果为(cm-1): 3078, 1602, 1058, 1336, 1134, 826, 620, 490。比较配体6与配合物14的红外光谱, 发现有较大的差异, 配合物的C=N伸缩振动吸收峰出现在1602 cm-1, 与配体相应峰1622 cm-1相比, 向低频移动了20 cm-1, 说明N原子参与了配位。配合物中的-OH特征吸收峰消失也说明了配合物的合成过程中, 羟基上的氢脱去。
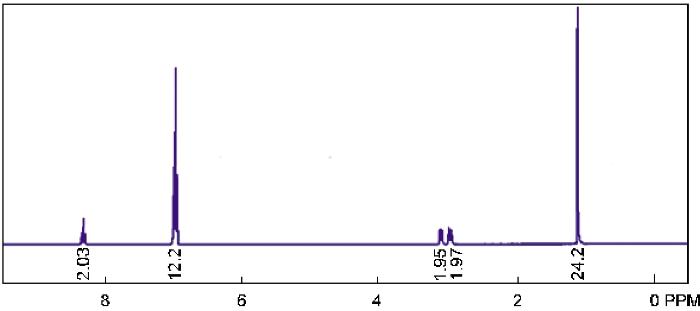
配合物15的1H-NMR表征结果, 如图5所示。从图5可知: δ= 1.17 (d, JHH=4 Hz, 24H, i-Pr-CH3), 2.92 (m, 4H, i-Pr-CH), 7.20 (s, 6H, Ar-H), 7.44 (s, 2H, Ar-H), 7.80 (s, 2H, Ar-H), 8.20 (s, 2H, CH=N)。元素分析(C38H40Br4O2N2TiCl2, 995.15 ), 实测值(计算值)(%): C 45.97 (45.86), H 4.22 (4.05), N 2.63 (2.81)。
配合物16的1H-NMR分析结果为: δ=1.22 (d, JHH=4.8 Hz, 24H, i-Pr-CH3), 1.31 (s, 18H, -C(CH3)3), 1.35 (s, 18H, -C(CH3)3), 2.98-3.29 (m, 4H, i-Pr-CH), 7.11-7.42 (m, 10H, Ar-H), 8.15 (s, 2H, CH=N)。元素分析(C54H76O2N2TiCl2, 903.99 ), 实测值(计算值) (%): C 71.98 (71.75), H 8.53 (8.47), N 3.02 (3.10)。
分别比较5与13, 6与14, 7与15, 8与16的1H-NMR数据, 可以看出: 配合物13、14、 15和16中的-OH峰消失。另外, 与O和N两配位原子较近的氢都发生了化学位移(i-Pr-CH, Ar-CH=N-向低场移动) [15, 16], 说明配体5 、6、7和8与金属钛化合物分别生成了配合物13、14、15和16。元素分析结果证实了目标化合物的组成。
在反应温度为60℃、反应压力2.0 MPa、反应时间为1 h的条件下, 配合物13、14、15和16分别催化乙烯聚合, 根据干燥后得到的聚合物产量计算了催化聚合活性; 用粘度法测定了聚乙烯样品分子量, 根据公式(1)计算的结果列于表1。
由表1中entries 1和3、1和5、1和7、1和9对比可知, 在无主催化剂的条件下助催化剂对乙烯聚合没有催化活性; 对比具有最高活性的entries 6和7可知, 在没有助催化剂的条件下主催化剂对乙烯聚合无催化活性, 且该规律适用于配合物13、14和16。当主催化剂和催化剂按照一定配比组成的催化体系催化乙烯聚合, 活性较高, 其主要原因是: 一方面, 助催化剂MAO可清除聚合体系中的有害杂质; 另一方面, 助催化剂MAO使卤代金属配合物烷基化并与主催化剂(卤代金属配合物)可形成真正活性中心, 维持活性中心的稳定性[21]。
表1的结果显示: 在相同的反应条件下, 取代苯氧基亚胺钛配合物14-16催化乙烯聚合活性的大小关系为: 15>14>16, 均比未取代苯氧基亚胺钛配合物13的催化活性(120.45 gPE /(mmolTihMPa))高得多。一种可能的解释是, 硝基、卤素等吸电子取代基的引入导致聚合活性中心数的大幅增加[14]。即配体上取代基吸电子增加了金属-碳阳离子活性中心数目, 导致乙烯插入活化能的降低, 且吸电子能力越强催化活性越高 [13, 14]。在苯氧基结构中引入较大的取代基如叔丁基(配合物16)也使配合物的催化聚合活性比无取代配合物大大提高。但是, 这类叔丁基取代配合物的合成比溴代或硝基取代配合物困难得多。因此, 配位催化剂15具有更大开发潜力, 而且与苯氧基亚胺配体上引入强吸电子的氟基F(s)和三氟甲基CF3(s)的三井化学Fujita催化剂[13, 14]相比, 具有较大的成本优势。
表1 配合物13-16催化乙烯聚合结果
Table 1 Ethylene polymerization for complexes 13-16
| Entry | Complex | PE(g) | Activity (gPE/(mmolTihMPa)) | Mv | M.P(℃)d |
|---|---|---|---|---|---|
| 1a | — | 0 | 0 | — | — |
| 3c | 13 | 0.53 | 120.45 | 19408 | 128.77 |
| 5c | 14 | 5.42 | 1231.82 | 45226 | 133.82 |
| 6b | 15 | 0 | 0 | — | — |
| 7c | 15 | 5.73 | 1302.27 | 35957 | 132.64 |
| 9c | 16 | 4.50 | 1022.73 | 28902 | 131.49 |
为了考察配合物催化乙烯聚合的活性寿命, 将配合物13-16分别在15、30、45、60、75、90、105和120 min终止聚合反应并计量聚合量。
从表2可见, 将称量的聚合物产量除以催化剂摩尔数、反应时间和聚合压力, 则随着反应时间的延长, 催化剂的活性先上升后下降, 在约60-75 min时到达最高活性值, 而120 min时, 基本上都已经失活了。这说明, 这类催化剂的活性寿命小于2 h。其主要原因是, 由于刚开始聚合时催化剂能被MAO充分活化, 表现出较高的活性; 随着聚合时间的延长聚合物的产量不断增加, 越来越多的催化剂颗粒被产物聚乙烯包裹, 失去了与乙烯单体接触的机会。此外, 反应后期聚合物粘度提高也减弱了活性中心与乙烯单体的充分接触, 在一定时间后链增长反应大大减弱[22, 23]。
表2 反应时间对乙烯聚合的影响
Table 2 Effect of reaction time on the ethylene polymerization
| Time / min | Polymer yield / g | |||
|---|---|---|---|---|
| Complex 13 | Complex 14 | Complex 15 | Complex 16 | |
| 15 | 0.10 | 1.24 | 1.34 | 1.05 |
| 30 | 0.23 | 2.63 | 2.77 | 2.13 |
| 45 | 0.39 | 4.05 | 4.28 | 3.30 |
| 60 | 0.53 | 5.42 | 5.73 | 4.50 |
| 75 | 0.59 | 6.50 | 6.84 | 5.44 |
| 90 | 0.60 | 7.55 | 8.03 | 6.31 |
| 105 | 0.61 | 7.79 | 8.16 | 6.42 |
| 120 | 0.61 | 7.80 | 8.17 | 6.44 |
比较图中各个配合物对乙烯聚合的影响可以发现, 四种配合物的活性高低依次为配合物15>配合物14>配合物16>配合物13。
表3列出了由凝胶色谱(GPC)测定的聚乙烯分子量及其分布。表中Mw为重均分子量; Mv为粘均分子量; Mp为峰值分子量; Mn为数均分子量; Mz为质均分子量; Mw /Mn为分子量分布。可以看出, 三种取代水杨醛亚胺钛化合物14/MAO、15/MAO和16/MAO体系催化得到的聚乙烯分子量高于未取代水杨醛亚胺钛化合物13/MAO, 分子量的分布则较窄, 具有单活性中心特征。主要原因是: 配合物在助催化剂作用下形成金属活性中心M[+]L[-], 从而引发乙烯聚合反应, 乙烯单体插入ML间完成链引发, 生成MCH2CH2[+]L[-]。链增长速率Rp={MCH2CH2L}{CH2=CH2}, 当链引发段浓度和乙烯单体浓度均较大时链增长速率较快, 能形成长链聚乙烯链段。配体L上吸电子的硝基或溴的存在增大了活性中性的正电性, 促进链引发进而增加Rp, 增大了聚合物链段。另外, 苯氧基的邻位有大的取代基团(叔丁基)时空间效应的存在能阻止活性中心与单体以外的其余组份的结合, 从而使配合物16形成的催化体系比无取代基的配合物13/MAO催化体系在聚合条件下保持更长时间的活性。于是, 催化所得的聚乙烯分子量较高, 分布也稍宽[24]。
表3 GPC c聚合表征结果
Table 3 Polymer characterization results determined by GPC c
| Complex | Mw | Mv | Mp | Mn | Mz | Mw /Mn |
|---|---|---|---|---|---|---|
| 13 | 20879 | 19266 | 18146 | 11079 | 32943 | 1.88 |
| 14 | 49319 | 44754 | 41937 | 23252 | 86256 | 2.12 |
| 15 | 38515 | 35108 | 33201 | 18434 | 65753 | 2.09 |
| 16 | 29677 | 27339 | 27116 | 15153 | 47093 | 1.96 |
尽管如上所述电子效应和空间效应使14-16催化体系引发乙烯聚合得到较高的分子量PE, 但是催化剂分子失活(分子失活包括活性链分子间的耦合反应和活性物种浓度高时分子间的耦合)和链转移反应(链转移反应包括: 聚合链向溶剂、杂质等分子的转移)使部分聚合链在未形成较大分子量时链增长反应即终止, 使聚乙烯分子量的分布较宽。进一步修饰配体的分子结构, 如苯氧基的对位引入强吸电子基团且其邻位引入阻止失活和链转移的大取代基, 则可能进一步调控聚合物的分子量和分子量的分布。
聚乙烯样品DSC测试结果, 如图6所示。从图6中四种PE样品(分别标记为13#、14#、15#、16#)的DSC测试结果可见, 14#、15#、16# 聚乙烯的熔融峰差别较小, 其熔点较高(大于130℃), 其中15#样品熔点高达132.6℃, 符合结晶聚乙烯的热性能特征。样品13#的熔点稍低, 其加工温度比其它样品加工温度低。
在同等条件下催化剂15催化乙烯聚合得到的聚乙烯活性最高, 达到了1302.27 gPE/(mmolTihMPa)。将催化剂15催化得到的聚乙烯样品进行13C-NMR表征, 结果如图7所示。
图7 配合物15在MAO催化体系下的聚乙烯产品的碳谱图
Fig.7 13C-NMR of PE produced by 15/MAO catalyst system
在图7中, 化学位移128-133 PPM处的三个峰是溶解PE所采用的氘代邻二氯苯溶剂所属的三种碳(1, 1位碳; 2, 2位碳和3, 3位碳)的谱线; 化学位移30 PPM处为线性聚乙烯链节-[CH2]n-的碳谱峰[25, 26]。聚乙烯样品的13C-NMR表征结果表明, 催化剂15所得聚乙烯具有饱和链端, 其原因是聚乙烯分子量较高及β-H转移反应受到限制[27]。此外, 聚乙烯样品的13C-NMR和DSC表征结果还表明, 催化剂15催化所得聚乙烯为线性结晶聚乙烯[6]。
在较高温度和较低MAO/cat摩尔比条件下, 取代水杨醛亚胺钛系催化剂14-16能有效催化乙烯聚合。在2.0 MPa, 60℃, nAl / nTi =1500∶1等工艺条件下聚合活性可达1022.73-1302.27 gPE /(mmolTihMPa), 均远高于无取代水杨醛亚胺钛二氯化物13。在配体中引入强吸电子的硝基和卤素基团, 可大大提高其催化乙烯聚合活性。用钛配合物15在MAO活化后甲苯溶液中催化乙烯均聚, 可得到高分子量线型结晶聚乙烯。强吸电子取代水杨醛缩苯胺类钛配合物14和15具有成本低、合成收率高、催化活性及聚合产物分子量高的特点, 工业开发的前景较大。

/
| 〈 |
|
〉 |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}