
熊需海 , 卢放
, 卢放
XIONG Xuhai, LU Fang
中图分类号: TB324, TQ323
通讯作者:
收稿日期: 2014-01-14
修回日期: 2014-05-6
网络出版日期: --
版权声明: 2014 《材料研究学报》编辑部 版权所有 2014, 材料研究学报编辑部。使用时,请务必标明出处。
基金资助:
作者简介:
展开
摘要
以酚酞型双马来酰亚胺(PBMI)和芳香二胺(DDE)为原料, 经迈克尔加成反应制备出新型环氧树脂芳胺固化剂(PBMI-DDE), 红外光谱和核磁共振等分析方法证实了新型固化剂的分子结构。用动态差示扫描量热法(DSC)确定了该固化剂和环氧树脂的固化工艺并对固化动力学进行了研究。采用Kissinger方程、FWO方程计算出固化反应活化能分别为 46.7 kJ/mol, 49.1 kJ/mol, 并结合Crane方程计算出反应级数为0.88。动态热机械分析表明, 完全固化的树脂体系玻璃化转变温度为153℃; 热失重研究结果显示, 新型环氧树脂体系具有优异的热稳定性。
关键词:
Abstract
A new curing agent (PBMI-DDE) for epoxy resin was synthesized from phthalide-containing bismaleimide (PBMI) and aromatic amine (DDE) according to the Michael addition reaction. The chemical structure of PBMI-DDE was characterized by 1H NMR spectroscopy and Fourier transform infrared spectroscopy (FTIR). The curing performance and kinetics of E-51/ PBMI-DDE was studied by DSC. Apparent activation energy evaluated according to Kissinger model or FWO model is 46.7 or 49.1 kJ/mol, respectively, and the calculated reaction order is 0.88 based on Crane method. The DMA and TGA results show that the cured resin possesses excellent thermostability with a glass transition temperature 153℃ for the completely cured resin.
Keywords:
环氧树脂(EP)是一种高性能热固性树脂, 具有粘接性能好、力学性能优异、耐腐蚀、抗疲劳等特点, 在电子电气、航空航天等诸多领域得到了广泛的应用。使用环氧树脂时必须加入固化剂, 固化反应后生成三维网状结构[1]。与酸酐类和咪唑类等中温固化剂相比, 芳香族胺类在固化过程中产生较多羟基, 有利于提高粘接性能, 广泛用作纤维增强复合材料的基体。但是, 芳香胺固化的环氧树脂的脆性大和耐湿热性能不佳。其原因是, 普通芳香二胺分子链较短导致固化物的交联密度以及单位体积内羟基的含量过高。因此, 改善环氧树脂的性能, 除了提高环氧树脂的质量、开发新型结构的环氧树脂之外, 合成新型结构固化剂也极为重要[2, 3]。
在环氧/芳胺树脂体系中加入双马来酰亚胺树脂(BMI), 能显著改善树脂体系的耐热性能和耐湿热性能[4-8]。但是, BMI树脂的溶解性较差导致改性树脂的溶液易析出BMI单体、存储性能不佳。另外, BMI的固化机制不同于环氧树脂, 改性树脂必须在高温下长时间固化才能保证BMI反应完全。设计合成含有BMI结构的芳香胺固化剂, 是解决上述问题的有效途径。BMI与芳香二胺的Michael加成物具有良好的溶解性, 能保证树脂溶液的稳定; 在固化过程中BMI不直接参与反应, 可采用传统环氧/芳胺树脂体系的固化工艺进行固化 [2]。此外, BMI结构的引入增加了固化剂的极性, 提高了树脂的内聚强度使粘接性能得到改善; 固化剂分子链段的延长能降低固化物的交联密度, 增韧环氧树脂; 刚性BMI分子链段的存在, 也能赋予固化物较好的耐热性能。
研究热固性树脂固化动力学, 有助于更好地了解树脂的性质及固化过程中的演变、确定合理的固化工艺, 从而使固化物性能最优化。本文采用新型含酞侧基链延长型双马来酰亚胺(PBMI)与芳香族二胺(DDE)反应制备链延长型芳香族二胺固化剂(PBMI-DDE), 用非等温DSC扫描法研究EP/PBMI-DDE体系的固化工艺、固化动力学, 并采用动态热机械分析和热重分析法表征其耐热性和热稳定性。
实验原料及纯化方法: 环氧树脂(E-51)(环氧值: 0.48~0.54, 工业品)、4, 4’-二氨基二苯醚(DDE)(化学纯)、丙酮(分析纯)、含酞侧基双马来酰亚胺(PBMI)(自制)[9]。丙酮先除水, 用KMnO4回流2 h, 常压蒸馏; 采用硅胶柱层析法分离提纯PBMI(流动相: 二氯甲烷/石油醚=3/1)。
测试仪器及方法: Nicolet 560 傅立叶变换红外光谱仪, KBr 涂膜法; Varian INOVA(400M)核磁共振仪, 氘代氯仿为溶剂, TMS为内标; XT-4型双目显微熔点测试仪; Netzsch公司DSC204差热扫描量热仪, N2氛围; PerkinElmer公司TGA-7型热重分析仪, N2氛围, 升温速率为20℃/min; TA公司的Q800 DMA, 样品尺寸为20 mm×6 mm×2 mm, 单悬臂梁模式, 驱动频率1.0 Hz, 升温速率3℃/min。
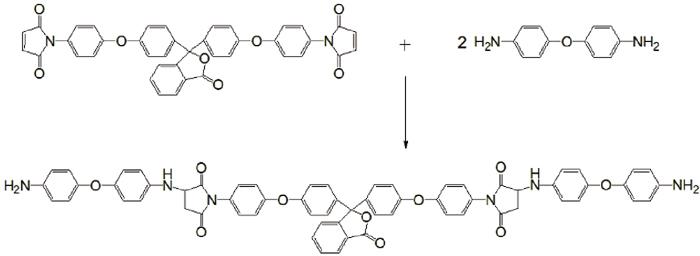
PBMI-DDE固化剂的合成: 先将0.2 mol DDE、250 ml丙酮及1 mL醋酸加入反应釜中, 在65℃下搅拌至DDE完全溶解, 然后分批加入0.1 mol PBMI, 反应3 h后体系变成均相透明溶液, 用薄层层析法检测反应终点(展开剂∶丙酮/石油醚=1∶1)。当PBMI的点消失后, 继续反应1 h以保证反应完全。把反应液冷却后倾入乙醇中, 过滤析出产物后用乙醇冲洗数遍, 放入80℃真空烘箱烘干, 得到产物104 g, 产率为97%, 熔点118~120℃。具体反应过程如图1所示。
E-51/PBMI-DDE树脂体系及固化物的制备: 将一定量的PBMI-DDE加入丙酮中, 搅拌至均相; 然后加入化学计量比的E-51, 继续搅拌10 min后在50℃真空脱去溶剂, 得到混合良好的E-51/PBMI-DDE树脂, 放入冰箱冷冻保存。将E-51/PBMI-DDE树脂放入涂有脱模剂的模具中, 加热到90℃后抽真空脱气, 然后按100℃×1 h→120℃×2 h→170℃×5 h程序温度固化, 自然冷却、脱模后得到固化物。
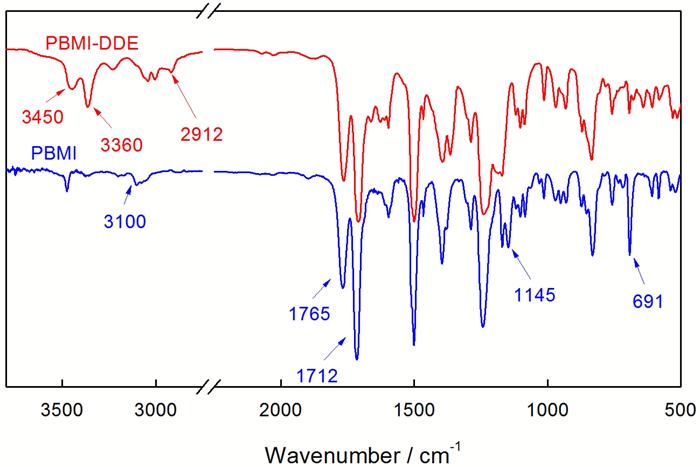
图2给出了PBMI-DDE, PBMI的红外光谱图。与PBMI的IR谱图相比, 在PBMI-DDE谱图中3100 cm-1附近的双键上的C-H伸缩振动峰、690 cm-1附近双键的伸缩振动吸收峰以及1145 cm-1附近马来酰亚胺环上C-N-C伸缩振动吸收峰等的消失了。这说明双马来酰亚胺参与了反应; 而在3450, 3360 cm-1处出现的伯胺(-NH2)的特征吸收峰以及2900~3000 cm-1范围内出现了-CH2-和-CH-的伸缩振动吸收峰, 证实了目标产物的生成; 另外, 在1710~1760 cm-1处两组羰基吸收峰没有变化, 说明反应过程中马来酰亚胺环和芳内酯环没有被破坏。
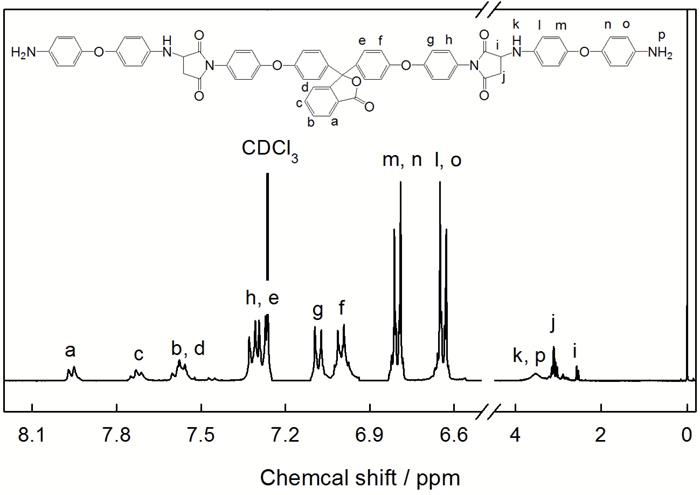
为了进一步验证PBMI-DDE的结构, 对其做了核磁共振氢谱分析。图3给出了PBMI-DDE的1H NMR及相应的归属。从图3可见, 谱图中没有明显的杂峰。在δ=6.85处马来酰亚胺环上双键相连氢的吸收峰消失了, 而在高场出现了饱和烃基和氨基氢质子的吸收峰; 综合考虑各个苯环氢质子吸收峰的强度比, 可确定反应产物是PBMI-DDE。
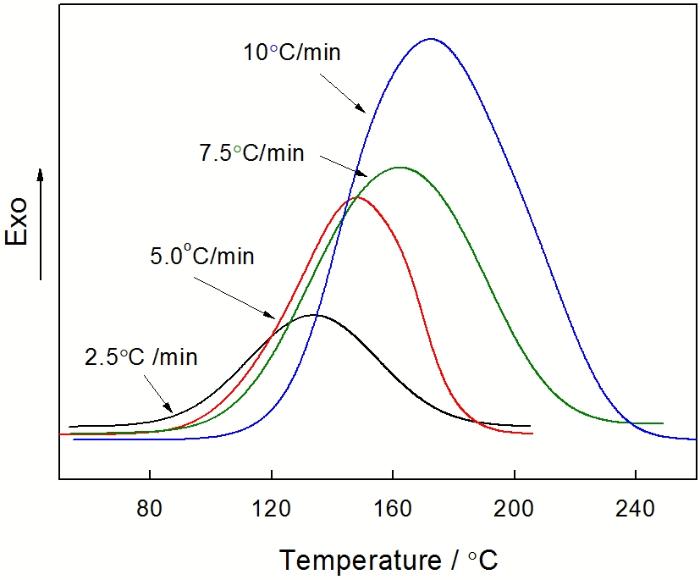
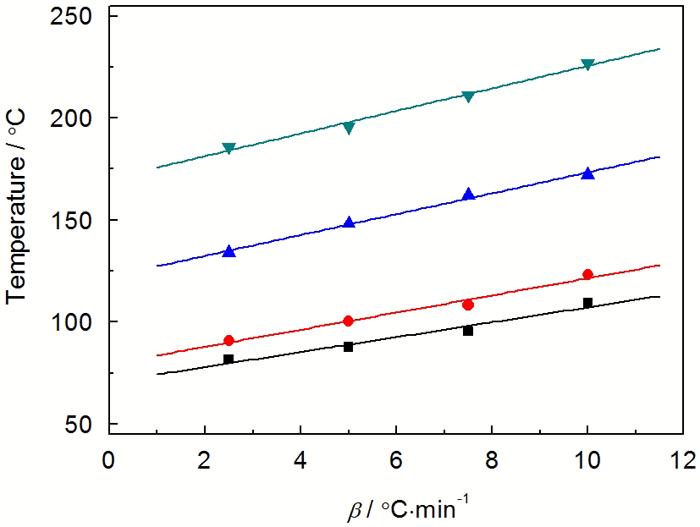
图4给出了采用非等温多重扫描得到的 DSC 曲线。由图4可以看出, 不同升温速率的DSC曲线都显示单一的放热峰, 且固化反应放热峰随着升温速率的增大, 固化放热峰向高温方向移动。这是因为, 升温速率增加则单位时间内产生的热效应增大、产生的温度差增大, 固化反应的放热峰相应地向高温方向移动, 固化反应加快。表1列出不同升温速率(β)下固化放热峰的特征参数, 其中Ti为起始固化温度、 Te 为外推起始固化温度、Tp为峰顶固化温度、Tt为终止固化温度、△H为固化热。
图4 E-51/PBMI-DDE体系在不同升温速率下的DSC曲线
Fig.4 DSC curves of E-51/ PBMI-DDE system at different heating rates
表1 E-51/ PBMI-DDE体系在不同升温速率下固化反应参数
Table 1 Curing characteristics of E-51/ PBMI-DDE systems at different heating rates
| β /℃min-1 | Ti/℃ | Te/℃ | Tp/℃ | Tt/℃ | △H /Jg-1 |
|---|---|---|---|---|---|
| 2.5 | 81.6 | 90.9 | 134.2 | 186 | 143.8 |
| 5 | 87.7 | 100.5 | 148.3 | 195.6 | 133.2 |
| 7.5 | 95.3 | 108.2 | 162.2 | 211.2 | 130.5 |
| 10 | 109.6 | 123.4 | 172.3 | 227 | 125.9 |
由图5可见, 外推至β=0时的温度参数Ti、Te、Tp和Tt分别为71、79、122和170℃。PBMI-DDE的分子链较为僵硬, 导致树脂体系在低温粘度较大、反应速度缓慢, 故应采用较高初始固化温度; 另外, PBMI-DDE的分子量较大, 单位体积内反应活性点较少且移动比较困难, 为确保树脂体系固化完全应适当延长高温固化时间。综合上述各种因素, E-51/PBMI-DDE体系的固化工艺确定为: 100℃×1 h→120℃×2 h→170℃×5 h。
表观活化能和反应级数是两个非常重要的固化动力学参数。根据表观活化能的大小可判断固化反应的难易程度。固化体系只有得到大于表观活化能的能量, 固化反应才能进行; 而根据反应级数则可分析固化反应的机理。
使用Kissinger方程和Flynn-Wall-Ozawa (FWO)方程可计算固化反应表观活化能[5, 10]。Kissinger法是一种用微分法对热分析曲线进行动力学分析的方法, 假设在固化过程中反应级数不变且固化反应的速率与DSC检测出的热流成正比, 即最大反应速率发生在固化反应放热峰峰顶温度处。该方法利用峰顶温度与升温速率之间的关系求解固化反应的表观活化能等动力学参数。
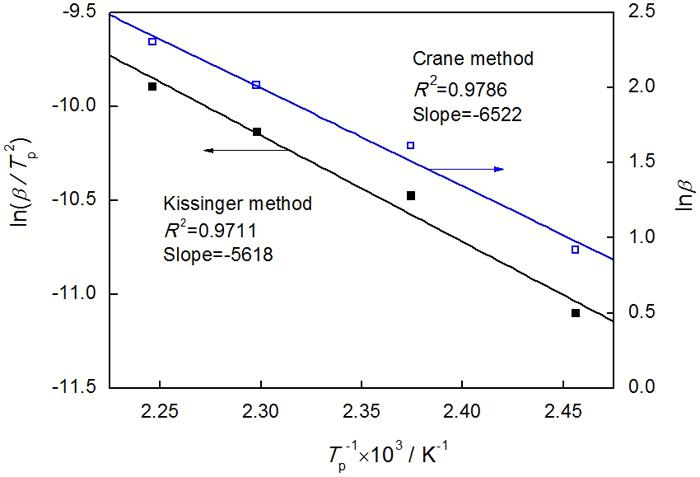
根据Kissinger 方程, 反应表观活化能Ea、固化反应放热峰值温度Tp与升温速率β之间的关系可以表达为
式中R为理想气体常数, A为频率因子。
以ln(β/TP2)对1/Tp作图, 如图6所示。由图6可以看出, ln(β/TP2)与1/Tp具有明显的线性关系。通过线性回归由斜率求得固化反应的表观活化能(Ea)为46.7 kJ/mol。
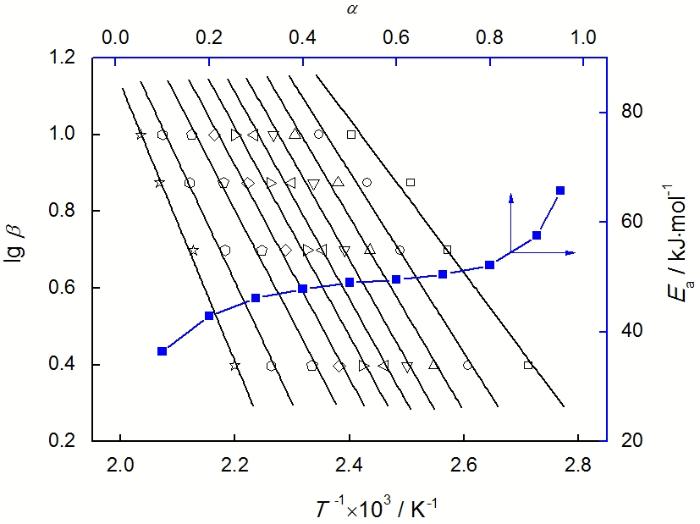
用Kissinger法计算表观活化能方便快捷, 但也有一定的局限性。特别是对于一些固化机理复杂的体系, 很难保证在整个固化过程中表观活化能不发生变化。 FWO模型避开了动力学机理函数直接求活化能, 避免了因反应机理的不同带来的误差[10]。利用等转化率下特征温度与升温速率的关系计算活化能, 能较全面地反映固化过程各个阶段表观活化能的变化。
根据FWO方程, 反应表观活化能Ea、特定转化率α对应的温度T与升温速率β之间的关系可以表达为
由于不同升温速率β对应的转化率α是特定值, 故积分形式的动力学机理函数G(α)是恒定的。
在特定转化率下lgβ对1/ T 作图, 由线性回归的斜率可求表观活化能Ea。图7给出了FWO法分析的结果。由图7可知, 固化反应在选定的固化区间(α=0.1~0.95)呈现出良好的拟合效果, 线性回归系数都在0.98以上。表观活化能随着固化反应的进行逐渐增大, 由初始阶段(α= 0.1)的36.4 kJ/mol增加到后期阶段(α= 0.95)的65.8 kJ/mol。其中在α<0.3和α> 0.8的范围内, Ea快速增加; 而在0.3<α<0.8区间内, Ea基本保持不变, 在整个固化过程中平均Ea约为49.8 kJ/mol。若以α= 0.5处的Ea作为整个固化过程的结果, 则E-51/PBMI-DDE体系的Ea近似值为49.1 kJ/mol, 与Kissinger法计算的结果相近。与其它含有Cardo或芳杂环结构的环氧/芳香二胺树脂体系相比[11, 12], E-51/PBMI-DDE体系的活化能处于较低的水平, 说明PBMI结构的引入并没有改变体系的固化反应活性。
使用Crane方程是求解反应级数的有效方法, 其与Kissinger法一样也是利用了DSC曲线的峰值温度 TP 与升温速率β的关系求解反应级数[11]。
根据Crane方程, Ea, TP, β与反应级数n的关系可以表达为
对于热固性树脂Ea/nR>> 2Tp, 式(3)简化为
对E-51/ PBMI-DDE体系, Ea取Kissinger法和FWO法求得表观活化能的平均值, 用lnβ对1/Tp作图(图6), 由直线的斜率即可求出n为0.88。由此可知, 该体系为复杂的固化反应体系。
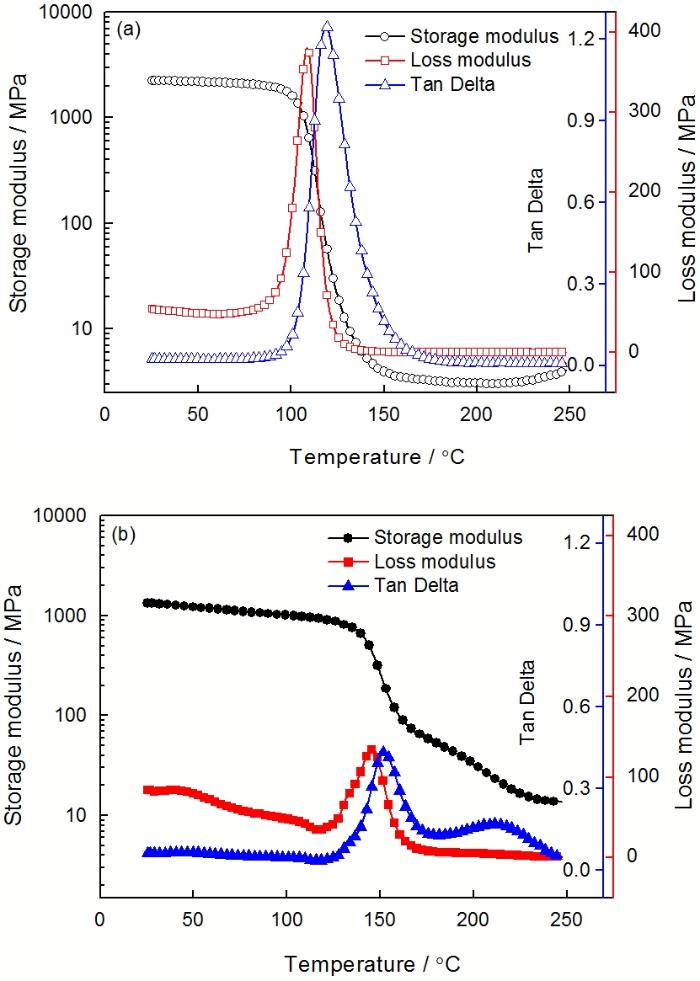
图8 E-51/ PBMI-DDE固化物的DMA曲线
Fig.8 DMA thermograph of cured E-51/ PBMI-DDE system.(a) the first scanning, (b) the second scanning
图8a, b分别给出了E-51/PBMI-DDE固化物的第一次扫描和第二次扫描的DMA曲线。从图8a中储能模量曲线可知, 固化物在玻璃态区域具有较高的刚度, 在100℃以下储能模量保持在2 GPa左右; 而在高弹态区域储能模量低于5 MPa。根据橡胶粘弹理论, E-51/ PBMI-DDE固化物的交联密度非常低。另外, 损耗模量峰值温度(109℃)和Tanδ峰值温度(120℃)均低于普通环氧/芳胺固化物的玻璃化转变温度, 也说明了固化物的交联密度不高。固化物的交联密度, 由树脂的结构和固化工艺决定。若固化不完全导致交联密度下降, 那么E-51/ PBMI-DDE固化物两次DMA扫描曲线则会有明显不同; 反之, 则可确定树脂结构是导致上述现象的主因。
比较图8a和8b发现, 第二次DMA测试的玻璃态储能模量约1.5 GPa, 低于第一次的测试值; 而高弹态储能模量介于20~100 MPa, 远高于第一次的测试结果。热固性树脂的玻璃态储能模量主要受分子链堆砌密度影响, 而橡胶态的储能模量主要与交联密度有关[13]。在第一次扫描过程中, 树脂体系中未反应的基团继续反应使固化物的交联密度得到提高; 而高温扫描也使分子链段的振动加剧, 自由体积增大并在测试结束时被“冷冻”从而导致堆砌密度下降。因此, 在第二次DMA扫描过程中出现了固化物玻璃态的储能模量下降而橡胶态的储能模量则升高的现象。另外, 图8b显示损耗模量峰值和Tanδ峰值明显低于第一次扫描的结果, 而其对应的温度则呈现相反趋势。这也说明, 经过第一次扫描后固化物交联密度增加。
由上述分析可知, DSC扫描外推法确定的固化工艺不能使E-51/ PBMI-DDE树脂体系完全固化, 为了提高耐热性必须经高温后固化处理。另一方面, 尽管E-51/ PBMI-DDE树脂固化物的交联密度低于普通芳胺固化物, 完全固化的树脂体系的耐热性与普通环氧/芳胺固化物的玻璃化转变温度相当。
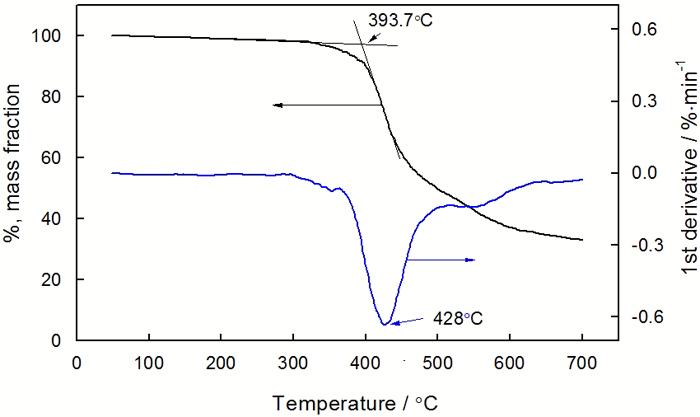
图9给出了 E-51/ PBMI-DDE固化物的 TG和 DTG曲线。TG曲线表明, 固化物在N2气氛中分解比较缓慢, 起始分解温度接近于400℃; 失重最快的温度为 428 ℃, 失重率为24.4%; 700 ℃时的残余重量达 33.2%。较高的残炭量主要归因于树脂体系中耐热芳杂环结构(酰亚胺环和内酯环)含量的增加 [14]。DTG曲线在主峰的高低温侧各有一个小峰, 说明固化物的分解过程是分步进行的。
图9 E-51/ PBMI-DDE固化物的TG和DTG曲线
Fig.9 TG and DTG curves of cured E-51/ PBMI-DDE system
通过PBMI和芳香二胺(DDE)的迈克尔加成反应, 可合成一种新型的环氧树脂固化剂(PBMI-DDE)。E-51 /PBMI-DDE体系的最佳固化工艺为 100℃×1 h→120℃×2 h→170℃×5 h。完全固化的E-51/PBMI-DDE体系的玻璃化转变温度为153℃, 与普通芳胺固化的环氧树脂相当。E-51/PBMI-DDE体系具有优异的热稳定性。

/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}