
邢佩 , 李晓刚
, 李晓刚
XING Pei, LI Xiaogang
中图分类号: TG172.4
文章编号: 1005-3093(2016)04-0241-07
通讯作者:
收稿日期: 2015-09-14
修回日期: 2015-09-30
网络出版日期: 2016-04-25
版权声明: 2016 《材料研究学报》编辑部 《材料研究学报》编辑部
基金资助:
展开
摘要
为了考察新型海洋平台用E690钢在海水环境中的耐蚀性能, 采用动电位极化曲线、电偶电流测量以及扫描电镜和拉曼光谱分析法研究了其在溶氧量为0.3-8 mg/L之间的3.5%NaCl溶液中的氧浓差腐蚀行为, 并讨论了溶氧量差值、阴阳极面积比以及锈层等因素对E690钢氧浓差腐蚀的影响。结果表明: 当阴阳极面积比Sc/a≤4时, 阴阳极面积比是影响氧浓差腐蚀的主要因素, 当Sc/a>4, 阴极区溶氧量是影响氧浓差腐蚀的主要因素; 锈层氧浓差腐蚀的影响与所处的环境有关, 处于贫氧条件下时, 对锈层下的金属有保护作用, 而处于富氧氧条件下, 锈层会参与阴极反应, 加速金属腐蚀, 并且由于锈层的不均匀性, 还会造成阳极金属的不均匀腐蚀, 产生点蚀坑。
关键词:
Abstract
In order to investigate the corrosion resistance of ocean platform steel E690 in sea water, the corrosion induced by oxygen-concentration cell of E690 steel in 3.5%NaCl with oxygen concentration within a range of 0.3 to 8 mg/L was investigated by means of electrochemical measurement techniques, scanning electron microscopy and Raman spectroscopy. The influence of the difference in dissolved oxygen, the ratio of cathode area to anode area and the corrosion product on the corrosion behavior of E690 steel was examined respectively. It was found that: when the ratio of cathode area to anode area was less than four (Sc/a≤4), the cathode and these ratios would be the main factor that influenced the oxygen-concentration cell corrosion; when Sc/a>4, the dissolved oxygen would be the main factor that influenced the oxygen-concentration cell corrosion; How the corrosion product influenced the oxygen-concentration cell corrosion depends on the dissloved oxygen. When the rusted metal under an oxygen-deficient condition, the rust layer would prevent the substrate from corrosion; when the rusted metal was immersed in an aerated condition, the corrosion product would participate in the cathodic reaction process, which would accelerate the anode dissolution and resulted in localized corrosion, such as pitting.
Keywords:
海洋是十分复杂的腐蚀性环境, 不同区域的海水溶氧量不尽相同[1], 海洋平台用钢贯穿海水的不同区域, 极易出现氧浓差腐蚀, 引发灾难性事故。多年来, 众多学者对氧浓差腐蚀进行了研究, 但都集中在土壤腐蚀中[2,3], 而对于海洋环境用钢的氧浓差腐蚀特征报道甚少[4]。谢建辉[5]以普通碳钢为实验材料研究了3.5% NaCl溶液中O2/ N2, O2 /空气和N2 /空气中3种氧浓差环境下金属的腐蚀强度变化。结果表明, 随氧浓度差异增大, 宏电池中阳极金属的腐蚀速率显著加速。在O2/ N2的环境条件下, 阳极金属的腐蚀速率比为构成氧浓差电池时的自然腐蚀速率大了7倍。以上研究中溶解氧浓度差异达到45 mg/L, 远远超出了海水中实际能达到的溶解氧浓差差异, 因此对于实际海洋环境中钢发生的氧浓差腐蚀行为借鉴意义不大。本文从实际工程使用环境出发, 研究海洋平台用E690高强钢在溶氧量在0.3-8 mg/L之间的3.5%NaCl溶液中的氧浓差腐蚀行为规律, 并讨论溶解氧浓差和阴阳极面积比两重因素共同作用时对氧浓差腐蚀的影响, 以及产生锈层后氧浓差腐蚀的变化。
实验基材为E690钢, 化学成分如表1所示。实验前工作电极用1500号砂纸研磨, 丙酮除油去污, 乙醇清洗后干燥备用, 以获得均匀光洁的的暴露表面。除工作面外, 试样四周用环氧树脂进行封装。
表1 实验用E690钢化学成分
Table 1 Chemical composition of experimental steels (%, mass fraction)
| C | Si | Mn | P | S | Alt | V | Cr | Ni | Cu | Mo | B | Ti |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.095 | 0.21 | 1.47 | 0.009 | 0.0012 | 0.0236 | 0.033 | 0.45 | 0.32 | 0.31 | 0.46 | 0.0018 | 0.015 |
实验采用E690钢制作试样, 其化学成分如表1。把试样打磨至2000#后抛光, 用4%(体积分数)硝酸酒精侵蚀, 分别用Polyvar MET金相显微镜观察其显微组织。
1.2.1 实验装置 电解质溶液为3.5%NaCl溶液, 采用蒸馏水和分析纯化学试剂配制而成。实验前向溶液中通N2除氧以控制溶液中的氧含量, 并用JPB-607A型溶氧仪测量溶液中的氧含量。
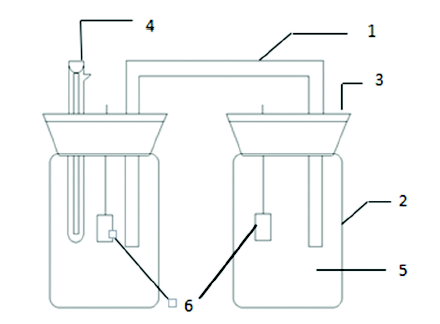
氧浓差电池实验装置如图1所示, 装置两侧的试样分别处于不同溶解氧含量的3.5%NaCl溶液中, 两侧溶液用盐桥连接, 将两试样用导线连接起来, 试样就处于短接状态, 组成了氧浓差电池。
图1 模拟氧浓差腐蚀的实验装置
Fig.1 The equipment of oxygen concentration corrosion cell simulation (1-salt bridge, 2-organic glass cylinder, 3-sealing plastics, 4-saturated calomel electrode, 5-3.5%NaCl, 6-E690 steel)
1.2.2 实验设置 改变装置两侧氧浓度梯度及阴阳极面积比, 研究其对E690裸钢的氧浓差腐蚀行为的影响。其中阳极室和阴极室中的溶解氧含量分别控制在0.3和2 mg/L、2和6 mg/L、0.3和6 mg/L以及0.3和8 mg/L。氧浓差电池中阳极试样的表面积为1 cm2, 阴极试样表面积分别取2、4和10 cm2, 厚度均为3 mm, 由此形成阴阳极面积比为1∶1、2∶1、4∶1和10∶1的腐蚀电池。
为了研究锈层的生成对氧浓差腐蚀的影响, 将带有预制锈层的E690试样与新鲜裸钢耦合浸泡在不同氧浓度的3.5%NaCl溶液中。并根据其开路电位的差异进行组合, 组成对应的氧浓差电池。其中, 预制锈层是将E690试样浸泡在3.5%NaCl溶液中采用恒电位(-0.3V vs. SCE)极化1 h获得的。
电化学极化和电化学阻抗谱测试在PGSTAT 302N电化学工作站系统上进行。电化学测量采用三电极体系, 工作电极为E690钢试样, 辅助电极为铂电极, 参比电极选用饱和甘汞电极(SCE)。测试前先将试样浸泡在溶液中15 min待开路电位稳定后再测量。极化曲线测试采用动电位扫描的方法, 扫描区间为开路电位±30 mV, 扫描速率为0.33 mV/s。
通过零电阻电流计耦接组成腐蚀电偶, 记录氧浓差电流。所有氧浓差电流和电位均使试样在各自溶液中稳定10 min之后再连接测量。
运用QUANTA250扫描电子显微镜(SEM)对除锈前后试样表面形貌进行观察, 采用LabRam HR Evolution Raman光谱对E690钢氧浓差电池腐蚀所产生的腐蚀产物成分以及预制锈层成分进行分析, 采用Ar+激光器输出的532 nm波长线作为激发线, 镜头为50倍长焦镜头, 光束直径为2 μm, 实测光谱范围为200-1500 cm-1。
E690钢原始试样金相组织如图2所示, E690钢组织主要由粒状贝氏体+少量板条状贝氏体组成, 组织相对均匀, 并没有明显的夹杂存在。
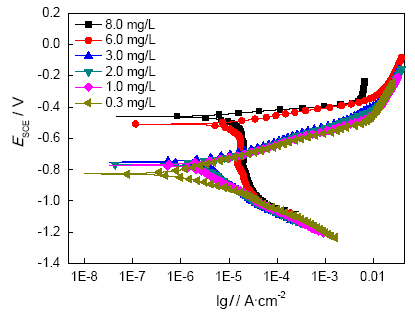
E690在不同氧含量的3.5%NaCl溶液中的动电位极化曲线如图3所示, 数据分析结果如表2。可以看出, 随着溶解氧含量的降低, E690的零电流电位逐渐降低, 腐蚀电流密度也显著降低。当溶氧量比较高时(6 mg/L, 8 mg/L), 阴极主要为吸氧反应, 阳极表现为活性溶解, 其中阴极反应为速控步。当溶解氧含量降低到3.0 mg/L时, 试样的零电流电位大幅下降, 电位下降到-0.8 V左右, 接近Q235钢的析氢电位[6], 此时溶氧量较少, 阴极反应由吸氧反应向析氢反应转变, 此时腐蚀电流也大幅降低, 小于10-6 A/cm2, 可以视作0。由上可知, O2是海水腐蚀中的去极化剂, 溶氧量越高, 阴极反应越快, 金属的腐蚀电流密度越大; 反之, 溶氧量的降低阻制了E690的腐蚀进程。
图3 E690在不同溶解氧的3.5%NaCl溶液中的动电位极化曲线
Fig.3 Potentiodynamic polarization curves of E690 in 3.5%NaCl with different DO (dissolved oxygen) concentration (20℃)
表2 3.5%NaCl中的电化学测试拟合结果
Table 2 Parameters of potentiodynamic polarization curves of E690 in 3.5%NaCl
| DO/(mg/L) | Ecorr/VSCE | Icorr/Acm-2 | Ba/(V/dec) | Bc/(V/dec) |
|---|---|---|---|---|
| 0.3 | -0.825 | 5.04×10-7 | 0.0319 | -0.3803 |
| 1 | -0.770 | 2×10-6 | 0.0473 | -0.2894 |
| 2 | -0.768 | 2.65×10-6 | 0.1017 | -0.2327 |
| 3 | -0.752 | 3.08×10-6 | 0.1100 | -0.2064 |
| 6 | -0.510 | 8.61×10-6 | 0.1002 | -0.1428 |
| 8 | -0.462 | 9.19×10-6 | 0.0828 | -0.0990 |
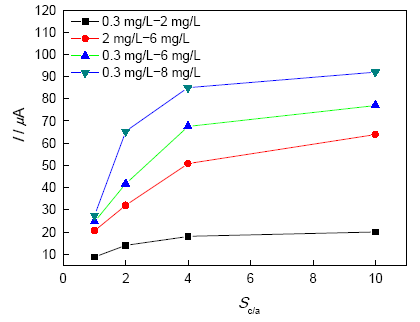
表3为不同氧浓差条件下E690钢的开路电位差, 图4为不同氧浓度差条件下E690不同阴阳极面积比组成宏电池时的电流值。可以看出, 随阴阳极氧浓差梯度的增加, E690钢发生氧浓差腐蚀的驱动力逐渐变大, 所产生的电流也不断增大。同一氧浓差条件下, 随着阴阳极面积比的增加, 电偶电流逐渐增大。但其增加速度逐渐减小。由此可知, 在该研究条件下, 氧浓差电流受氧浓度差异和阴阳极面积比的共同影响。当阴阳极面积小于等于4时, 氧浓差电流的增加主要由面积比的增加引起; 阴阳极面积比大于4后, 氧浓度梯度对氧浓差电流的影响占主要地位, 此时再增加阴阳极面积比, 电偶电流增加不大。
表3 氧浓差电池基本参数
Table 3 Parameters of the oxygen concentration corrosion cells
| DO in the two containers | Oxygen concentration difference | OCP difference |
|---|---|---|
| 0.3-2 mg/L | 1.7 mg/L | 54 mV |
| 2-6 mg/L | 4 mg/L | 95 mV |
| 0.3-6 mg/L | 5.7 mg/L | 149 mV |
| 0.3-8 mg/L | 7.7 mg/L | 170 mV |
图4 面积比对氧浓差腐蚀电流的影响
Fig.4 Influence of area ratio on oxygen concentration corrosion
由以上结果可以推测, 在3.5%NaCl溶液中, 氧浓差电池阴阳极面积比Sc/a≤4时, 阴极表面积相对较小, 消耗氧的速率有限, 扩散过程不是速控步, 因此此时的阴极反应主要取决于阴极表面在耦合电位时的氧还原反应速率, 此时增加阴极表面积, 为氧还原反应提供更多的活性表面, 氧浓差电流显著增加; 当Sc/a>4, 此时阴极表面积足够大, 消耗氧的速率也足够快, 阴极反应取决于阴极去极化剂氧的极限扩散电流, 此时加大阴极区域的溶氧量, 促进其向试样表面扩散, 使得氧浓差电流显著增加。
2.3.1 锈层对E690氧浓差腐蚀电化学行为的影响 对不同含氧量的3.5%NaCl溶液中的带锈金属以及新鲜裸钢进行开路电位监测, 结果见表4。对比分析后可知, 在贫氧溶液中带预制锈层的试样开路电位更正, 而新鲜表面试样在富氧环境中的电位更正。由此可以确定三组氧浓差电池, 如表5所示。图5为三组氧浓差电池的浓差电流随时间的变化。
表4 不同溶氧量下试样的开路电位
Table 4 OCP in 3.5%NaCl solution with different dissolved oxygen
| DO/(mg/L) | OCP/V | |
|---|---|---|
| Rusted sample | Unrusted sample | |
| 0.3 | -0.698 | -0.741 |
| 6.0 | -0.656 | -0.556 |
表5 三组氧浓差腐蚀电池的设置
Table 5 Setting of the three oxygen concentration corrosion cells
| The cathode and anode are respectively in each solution | The cathode and anode are in the same solution | |||
|---|---|---|---|---|
| Group one | Group two | Group three | ||
| Anode | Fresh steel (solution with 0.3 mg/L DO) | Rusty metal (solution with 0.3 mg/L DO) | Rusty metal (solution with 6.0 mg/L DO) | |
| Cathode | Rysty metal (solution with 6.0 mg/L DO) | Fresh steel (solution with 6.0 mg/L DO) | Fresh steel (solution with 6.0 mg/L DO) | |
图5 3组氧浓差电池电流和电位随时间的变化
Fig.5 Corrosion current-time curves of the oxygen concentration corrosion cell, (a) the first group, (b) the second group, (c) the third group
由图5及表4可知, 第一组氧浓差电池电位差为85 mV, 氧浓差电流先增大后减小, 到第7天后几乎减小到0。第二组氧浓差电池的电位差最大(相差142 mV), 但氧浓差电流却最小。第三组氧浓差电池, 电位差为100 mV, 但在实验过程中氧浓差电流一直保持较大值, 均在20 μA以上。
2.3.2 腐蚀形貌 三组氧浓差电池中的阳极除锈前后的表面形貌如图6所示。可以看到, 除锈后, 三组氧浓差电池的阳极表面都有明显的点蚀坑, 且点蚀坑的深度和数量与氧浓差电流的大小顺序一致, 即氧浓差电流越大, 点蚀坑的数量越多, 深度越深。未除锈前, 第二组氧浓差电池的阳极表面锈层最为均匀致密, 其他两组腐蚀电池的阳极锈层均出现明显的裂纹, 第三组浓差电池的阳极表面黑色的内锈层上面又长出一层较厚的黄色外锈层, 内锈层裂纹较深较宽, 外锈层疏松多孔。
图6 3组氧浓差电池阳极表面除锈前、后的腐蚀形貌
Fig.6 Corrosion morphology of the anode surface with rust layer (a, c, e) and without rust layer (b, d, f), (a, b) the first group, (c, d) the second group, (e, f) the third group
2.3.3 腐蚀产物分析 不同条件下的锈层成分检测结果如图7所示。
图7 试样表面锈层成分激光拉曼光谱
Fig.7 Raman spectra of the corrosion rusty layer, (a) E690 after potentiostatically anodic polarization, (b) corrosion products on the cathode surface of the first group of oxygen concentration corrosion cell, (c) prefabricated rust layer sample after immersed in solution with full dissolved oxygen
对照各标准物相(铁的氧化物和羟基氧化物)激光Raman光谱的特征峰位, 可推知不同条件下E690试样腐蚀产物的组成, 见表6。
表6 产物拉曼峰和标准特征峰位的对应表
Table 6 Raman peak frequencies of corrosion products and standard rust phase
| Raman peak frequencies of corrosion products / cm-1 | Raman peak frequencies of standard rust phase / cm-1 | Corrosion products | |
|---|---|---|---|
| Corrosion products made by polarization | 218, 286, 489, | 225, 245, 295, 415, 500, 615, | α-Fe2O3 |
| 328, 406 | 330, 415, 745 | β-FeOOH | |
| Corrosion products on the cathode surface of the oxygenconcentration corrosion cell | 319, 680 | 298, 319, 420, 560, 680, 1322 | Fe3O4 |
| Corrosion products after immersed in solution | 265, 345, 386, | 265, 300, 345, 395, 515, 645, 670, 715, | γ-Fe2O3 |
| 485, 680 | 248, 303, 397, 485, 554, 680, 1002 | α-FeOOH |
根据以上分析可知, 由恒电位极化预制的E690锈层其主要成分为β-FeOOH和α-Fe2O3。第一组氧浓差电池中, 带锈层金属试样做阴极时锈层的主要成分变为Fe3O4, 而带锈金属自然浸泡后, 锈层的主要成分为γ-Fe2O3、α-FeOOH。对比E690三种不同的腐蚀状态, 预制锈层及其自然浸泡后的成分均与组成氧浓差电池产生的锈层成分不同, 说明在氧浓差电流的作用下, 锈层进一步发生了不同于自然浸泡下发生的化学反应。
2.3.4 腐蚀过程分析 根据以上测试结果, 可推知第一组氧浓差腐蚀电池中(新鲜裸钢为阳极, 带锈金属为阴极)氧浓差电流随时间先增大后减小, 主要原因在于E690预制锈层中的成分发生转变, 如式(1)所示腐蚀开始时, 这一转变在一定程度上作为阴极反应参与了氧浓差腐蚀, 促进了腐蚀进程, 这时阴极可能发生两种还原反应(式1和2)。有关文献也证实β-FeOOH具有较强的还原性[7-10], 会发生还原反应, 转变为Fe3O4。
随着氧浓差腐蚀的进行, 做为阳极的裸钢表面出现锈层, 如图7a所示, 这在一定程度上阻碍了阳极反应的进行, 同时阴极表面锈层成分改变, β-FeOOH转变为晶粒较细并且更加稳定的Fe3O4[11], 使得锈层变得更加致密, 致密均匀的锈层阻隔了O2向基底金属传输, 阴极反应也受到阻碍, 此时阴阳极的电位差减小, 因此氧浓差电流减小。
第二组氧浓差电池(预制锈层金属为阳极, 新鲜裸钢为阴极)电位差高达140 mV, 比第一组氧浓差电池中的电位差(85 mV)高出55 mV, 但氧浓差电流最小, 这表明无氧条件下的锈层阻碍了Fe离子的溶解析出, 对基体有保护作用。同时, 作为阴极的新鲜裸钢处于富氧状态下, 自身会发生较快的腐蚀, 表面长出锈层, 阻碍了阴极去极化剂O2与金属表面的接触, 阴极反应也受到阻碍, 因此氧浓差电流迅速减小接近于0。
第三组氧浓差电池(预制锈层金属与新鲜裸钢处于富氧溶液中)电位差为100 mV, 氧浓差电流也较高, 一直保持在20 μA以上, 此时处于阳极的金属由于表面预制锈层中存在活性成分β-FeOOH, 易发生如式(1)所示的阴极反应, 此时除了氧浓差造成的腐蚀外, 阳极金属的溶解还受到锈层阴极反应的加速作用, 所以该组电池中阳极金属基体腐蚀最严重, 产生多且密的点蚀坑, 如图7f。
综合分析以上三组氧浓差腐蚀电池可知, 当锈层处于有氧条件下时, 氧浓差电流更大, 这是因为在有氧条件下, 预制锈层试样中含有的活性腐蚀产物, 例如β-FeOOH, 会发生化学转化, 这些反应都会产生电子的交换, 加速了溶液中离子的定向流动, 从而产生较大的氧浓差电流。而在无氧条件下, 预制锈层试样电位为-696 mV, 不在锈层中的活性成分β-FeOOH的还原电位-650— -690 mV[12]范围内, 此时锈层的覆盖只是起了屏蔽作用, 阻碍了金属表面腐蚀反应的进行, 减缓了氧浓差腐蚀。
1. 溶解氧含量的降低抑制了E690钢的阳极溶解, 并使E690钢的阴极反应由吸氧反应向析氢反应转变。当溶解氧含量低于3 mg/L时, E690钢的阴极反应变为析氢反应。
2. 在3.5%NaCl溶液中, 氧浓差电池阴阳极面积比Sc/a≤4时, 氧浓差电流大小取决于阴极表面在耦合电位时的阴极反应速率, 此时, 增加阴极表面积, 氧浓差电流显著增加; 当Sc/a>4时, 氧浓差电流大小取决于阴极去极化剂氧的极限扩散电流密度, 此时, 阴极溶解氧含量增加, 氧浓差电流密度增加不明显。
3. 溶液中的含氧量是锈层中的活性成分发生转变的决定性因素。锈层处于无氧条件下时, 其中的活性成分不会发生转变, 此时锈层对金属只有保护作用; 在有氧条件下, 锈层中的活性成分则会发生化学转变, 参与阴极反应, 从而加速氧浓差腐蚀。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
Macrocell corrosion of Q235 steel in polluted soils due to the difference of oxygen concentration ,Q235钢在污染土壤中的氧浓差宏电池研究 , |
| [3] |
Macro-cell corrosion of X70 steel between salinized soil and seawater ,X70钢的滨海盐渍土-海水宏电池腐蚀 ,
利用电化学阻抗测试技术等,研 究了X70钢在滨海盐渍土与海水构成的宏电池中的腐蚀行为。结果表明,构成宏电池之后,海水侧X70钢一直作为宏电池的阳极处于被腐蚀状态,其腐蚀速率为 自然腐蚀速率的25.3倍,腐蚀过程主要受阴极反应控制;由于腐蚀产物膜和氯离子的双重作用使宏电池腐蚀速率先有一定程度的降低,然后迅速升高,再有所降 低并趋于稳定。试验还发现土壤-海水构成的宏电池的电动势并没有表现出像常规土壤宏电池电动势那样的先升高后降低的趋势,而是先有一定程度的降低,然后迅 速升高后趋于稳定,该过程主要受极化电阻的影响。
|
| [4] |
The differential aeration cell and the corrosion paradox ,
Steel with a low volume of both anolyte and catholyte was used for monitoring of steel electrode corrosion rates under conditions in the environment of a differential aeration cell under the conditions of both stagnating and exchanging electrolyte. A 3% NaCl solution was used as the electrolyte. A low content of oxygen near the anode was ensured by purging the solution with nitrogen while a high content of oxygen near the cathode was obtained by saturating the solution with oxygen. All experiments were performed for 7265h. Corrosion rates of steel electrodes were identified by using the weight loss method in comparison with the resistometric method. The corrosion rate of an aerated electrode drops in stagnating electrolyte to a hundredth of millimetre per year (0.0165mm/a) as a result of the catholyte alkalization and subsequent surface passivation, and that it is lower than the corrosion rate of a non‐aerated electrode (0.1265mm/a). In a stagnating solution, the catholyte is alkalized (pH 11) whereas acidification of the anolyte is negligible. In the case of continuous exchange of the aerated catholyte and the oxygen‐depleted anolyte in the environment of a differential aeration cell, the cathode's corrosion rate is up to seven times higher (0.765mm/a) than the corrosion rate of the anode (0.165mm/a). The difference between the electrodes' corrosion rates is given by the IR drop in the electrolyte between the cathode and anode. It was experimentally proved that in the case of continuous electrolyte exchange in the differential aeration cell the cathode corrodes faster than the anode and that no corrosion paradox is observed.
|
| [5] |
Investigation on oxygen concentration difference macrocell corrosion for steel A3 ,A3钢的氧浓差宏电池腐蚀作用的研究 , |
| [6] |
Corrosion behavior of Q235 in simulated natural environment by electrochemical technology ,Q235钢在模拟自然环境下失效行为的电化学分析 , |
| [7] |
Failure analysis of a 300M steel pressure vessel ,
A failure analysis was made in a 300M steel pressure vessel which has failed during hydrotest. The rupture occurred suddenly at a pressure level lower than what has been expected for the proof pressure. According to the results of various examinations it was concluded that hydrogen assisted stress corrosion cracking was the mechanism responsible for the failure. The root causes of this failure have been identified, serving as base for the modification of testing procedures in similar components.
|
| [8] |
Corrosion of low carbon steel in atm ospheric environments of different chloride content ,
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">This investigation aims to analyze the effect of Cl<sup>−</sup> ion on the atmospheric corrosion rate of carbon steel. The metal samples were exposed to a marine atmospheric environment (95 and 375 m from the sea line) as well as an industrial atmospheric environment. The effects of Cl<sup>−</sup> ions on the protective characteristics of the rust layers were assessed by IR spectroscopy, SEM–EDAX analyses, linear polarization resistance and electrochemical impedance spectroscopy (EIS). The results show that Cl<sup>−</sup> ion influences the corrosion rate, as well as the morphology and composition of the rust layer.</p>
|
| [9] |
Formation of the Fe(II)-Fe(III) hydroxysulphate green rust during marine corrosion of steel ,
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">Rust layers formed on steel sheet piles immersed 1 m above the mud line for 25 years were analysed by Raman spectroscopy, scanning electron microscopy and elemental X-ray mappings (Fe, S, O). They consist of three main strata, the inner one mainly composed of magnetite, the intermediate one of iron(III) oxyhydroxides and the outer one of hydroxysulphate green rust GR(SO<sub>4</sub><sup>2−</sup>). Simulations of GRs formation in solutions having large [Cl<sup>−</sup>]/[SO<sub>4</sub><sup>2−</sup>] ratios revealed that the hydroxysulphate GR(SO<sub>4</sub><sup>2−</sup>) was obtained instead of the hydroxychloride GR(Cl<sup>−</sup>), as demonstrated by X-ray diffraction and transmission Mössbauer spectroscopy analyses. Measurements of the [S], [Fe] and [Cl] concentrations allowed us to establish that GR(SO<sub>4</sub><sup>2−</sup>) formed along with a drastic impoverishment of the solution in sulphate ions; the [Cl<sup>−</sup>]/[SO<sub>4</sub><sup>2−</sup>] ratio increased from 12 to 240. The GR, acting like a “sulphate pump”, may favour the colonisation of the rust layers by sulphate reducing bacteria.</p>
|
| [10] |
The long term growth of the protective rust layer formed on weathering steel by atmospheric corrosion during a quarter of a century , |
| [11] |
Advances in understanding atmospheric corrosion of iron II. Mechanistic modelling of wet-dry cycles ,
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">With the aim of predicting the long term atmospheric corrosion behaviour of iron, the mechanisms occurring inside the rust layer during a wet–dry cycle are considered as well as the characteristics of the rust layer formed during this process. A first step in modelling the behaviour is proposed, based on the description of the cathodic reactions associated with iron oxidation: reduction of a part of the rust layer (lepidocrocite) and reduction of dissolved oxygen on the rust layer. The blocking of the anodic sites is considered to describe the extinction of electrochemical corrosion during the drying. The modelling, by including some composition and morphological data of the rust layer as parameters is able to account for the metal damage after one wet–dry cycle.</p>
|
| [12] |
Electrochemical study of indoor atmospheric corrosion layers fromed on ancient iron artefacts ,
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">Several indoor atmospheric corrosion layers (0–800 years old) were selected from different localisations in France. Each sample was scrapped from its iron substrate. The resulting powder was mixed with graphite in appropriate proportions and the mixture was pressed onto a stainless steel grid to constitute a composite electrode. The electrochemical responses of the different samples were recorded under galvanostatic regulation, in a near-neutral pH-buffered NaCl solution at 25 °C. The <em>E</em>–<em>t</em> reduction curves allowed the determination of two characteristic parameters, <em>E</em><sub><em>τ</em>/2</sub>, the potential value obtained at half the transition time, and <em>Q</em><sub><em>τ</em></sub>, the coulombic charge obtained at the end of the reduction. The diminution of <em>E</em><sub><em>τ</em>/2</sub> and <em>Q</em><sub><em>τ</em></sub> with the age of the corrosion layer showed that the “reduction reactivity” decreases with time, suggesting a progressive stabilisation of the corrosion layer.</p><p id="">In a second part of the work, we synthesised several common ferric or ferrous/ferric products (goethite, lepidocrocite, magnetite, maghemite, ferrihydrite) and compared their reduction responses (product alone or mixture of 2 or 3 products) to those of corrosion samples.</p>
|

/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}