
彭晓 , 王福会
, 王福会
中国科学院金属研究所金属腐蚀与防护国家重点实验室, 沈阳110016
PENG Xiao, WANG Fuhui
中图分类号: TG172.5
通讯作者:
收稿日期: 2013-09-22
修回日期: 2013-09-22
网络出版日期: --
版权声明: 2014 《金属学报》编辑部 版权所有 2014, 金属学报编辑部。使用时,请务必标明出处。
基金资助:
作者简介:
彭 晓, 男, 1967年生, 研究员, 博士
展开
摘要
一些金属基结构材料, 不需要增加Cr和Al含量而只需“纳米晶化”, 就能够在高温环境下形成保护性Cr2O3或Al2O3氧化膜. 纳米晶化是施加高Cr高Al涂层之外提高金属材料抗高温腐蚀性能的另一途径. 近20年来, 纳米晶金属材料的高温腐蚀行为已广泛报道. 本文简要评述了纳米晶金属材料的高温腐蚀特性、纳米晶化提高金属抗氧化性能的根本原因以及亟待澄清的问题.
关键词:
Abstract
Some structural metallic materials, with only necessary to be “nanocrystallized” rather than to be increased with the contents of Cr and Al, have the ability to thermally grow a protective scale of Cr2O3 and Al2O3 at high temperature environments. Nanocrystallizationof alloys containing Cr or/and Al is an alternative to conventionally coating them with a high-Cr or/and high-Al material. High temperature corrosion behaviors of nanocrystalline metallic materials have been extensively reported in the past 20 years. In this paper, characteristics of high temperature corrosion of nanocrystalline metals, together with the fundamental reasons and questions desired to be clarified for the improvement of the corrosion resistance of alloys by nanocrystallization, were briefly reviewed.
Keywords:
大多数金属材料暴露在高温氧化环境中, 不可避免热生长氧化膜. 具有保护性的氧化膜必须致密、热稳定性佳、 生长速率慢. 常见保护性氧化膜为Al2O3和Cr2O3. 其中Cr2O3在低于1000 ℃和II型热腐蚀(700~800 ℃)条件下具有良好的抗高温腐蚀性能, 而Al2O3在高于1000 ℃和I型热腐蚀(800~950 ℃)条件下具有良好的抗高温腐蚀性能[
纳米晶化是区别于提高Cr和Al量而促使金属材料热生长保护性氧化膜的另一途径. Giggins和Pettit[
合金纳米晶化主要采用PVD和机械方法. 普通合金靶材通过PVD被蒸发或轰击出的原子在样品基体上沉积, 获得化学成分与母合金相似的纳米晶合金(垂直于沉积方向的为纳米等轴晶, 而平行于沉积方向的一般为柱状晶). 一些合金也可通过喷丸[
众所周知, 抗高温腐蚀性能主要取决于两方面: (1) 氧化膜生长(或与环境介质反应)速率; (2) 氧化膜与金属基体的界面结合力. 研究表明, 纳米晶合金与普通粗晶的相比抗高温腐蚀性能提高的原因在于: Cr和Al可选择性氧化形成Cr2O3或Al2O3膜, 且氧化膜黏附性好. 为此, 本文根据相关研究结果和进展, 就金属材料纳米晶化如何促进选择性氧化及提高氧化膜的黏附性进行简要评述.
纳米晶化的明显优势是促使非保护性氧化膜生长型的、含Cr或(和)Al低的金属材料发生选择性氧化, 从而显著降低其氧化速度. 比如, 对于Ni-Cr二元普通粗晶合金, 热生长Cr2O3膜所需的临界含量为20% (质量分数, 下同)[
基于Wanger理论[
式中,
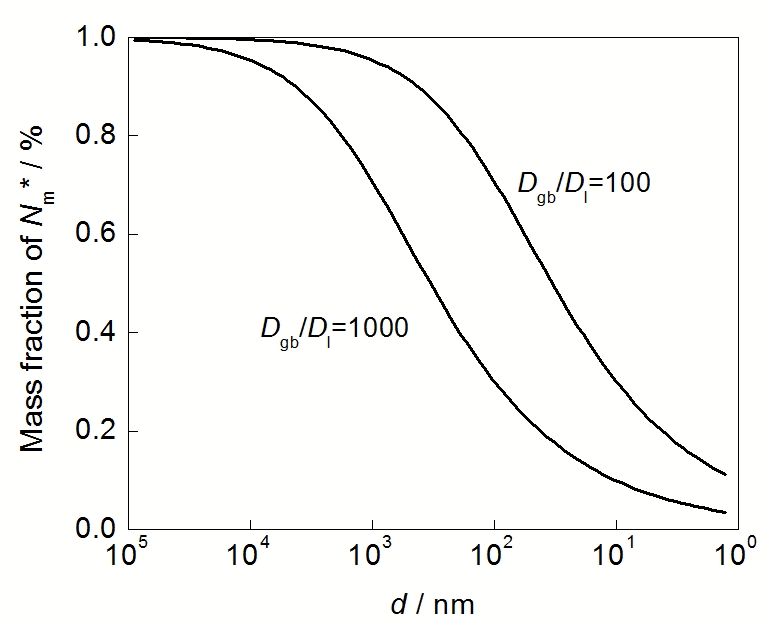
式中, ƒ为晶界体积分数. 若晶粒为简单立方形, 则ƒ=2δ/d (δ为晶界宽度, d为晶粒尺寸). 若Dgb>>Dl, 则由式(1)和(2), 可获得
式中,
对于多元多相合金体系, 成膜元素选择性氧化的临界含量计算比较复杂, 其在金属中的扩散还受到其它原子的作用, 但是这些体系晶粒细化尤其纳米晶化降低
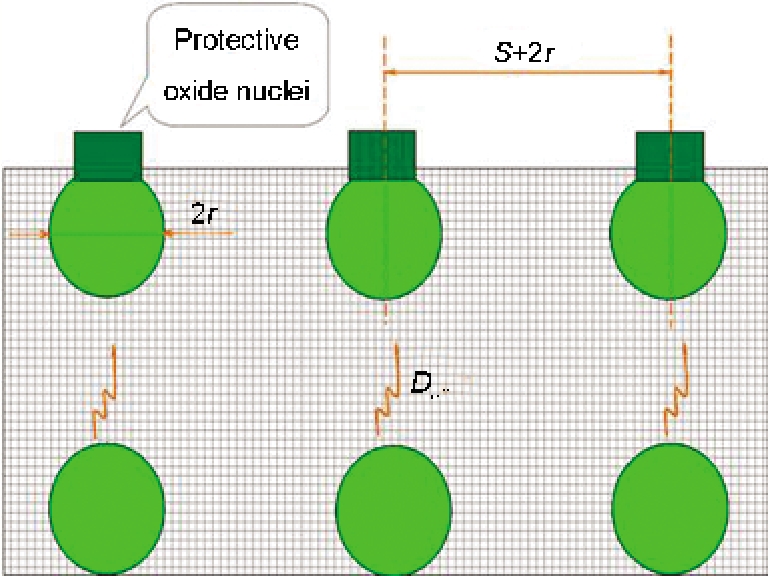
Wagner理论虽可解释纳米晶化促进选择性氧化的问题, 但并无助于深入认识纳米晶化促进选择性氧化的物理和化学过程. 金属氧化可分为2个阶段: 暂态(初期)氧化和稳态氧化. 以Ni-Cr二元合金为例, 暂态氧化是连续的Cr2O3膜的形成过程, 而稳态氧化是Cr2O3膜形成后的增厚过程. Ni-Cr合金的选择性氧化过程关键在于初期阶段. 基于纯金属初期氧化过程的认识[
综上所述, 一定氧化条件下的纳米晶合金的选择性氧化过程, 取决于2方面: 一是保护性氧化物形核密度高, 它主要受Cr和Al含量的影响; 二是该氧化物形核后的生长速度快, 它受Cr和Al在金属中的扩散速度影响. 上述Cr颗粒尺寸为21 nm的Ni-7.5Cr能发生Cr的选择性氧化, 而Cr颗粒尺寸为39 nm的只形成NiO膜, 就是由于Cr2O3的形核密度不够高、Cr2O3核在被NiO包裹之前不能连接起来的缘故. 如果弥散颗粒为微米级, 则Ni-Cr发生选择性氧化的
值得一提的是, 由于合金纳米晶化后改变了氧化物的形核和生长过程, 对于M-Cr-Al合金, 其纳米晶化, 也可使得Cr2O3膜生长型的合金转化为Al2O3膜的合金. 例如, K52合金(其中含22%Cr和3%Al)氧化一般热生长Cr2O3膜, 而其纳米晶化后却能生长Al2O3膜[
Cr2O3或Al2O3氧化膜一旦形成后, 氧化过程就由暂态阶段进入稳态阶段, 氧化动力学由阴、阳离子在氧化膜的扩散控制. 但是, Cr2O3或Al2O3膜能否持续生长, 取决于到达氧化膜/金属界面的Cr或Al原子通量(Jm)是否能平衡膜中离开界面的Cr或Al离子的通量(Jox). Jox大小与膜的生长动力学相关, 而Jm大小决定于成膜元素在合金中的浓度和有效扩散系数
式中, kc为氧化膜生长的抛物线常数. Cr2O3或Al2O3的kc值很低, 多数情况下远小于
虽然还缺乏相关合金纳米晶热生长的动力学数据, 但合金中的微量元素和杂质在晶界偏聚或析出会阻滞晶粒长大, 因而合金纳米晶即使粗化, 晶粒尺寸也会比普通合金的低得多, 导致成膜元素的
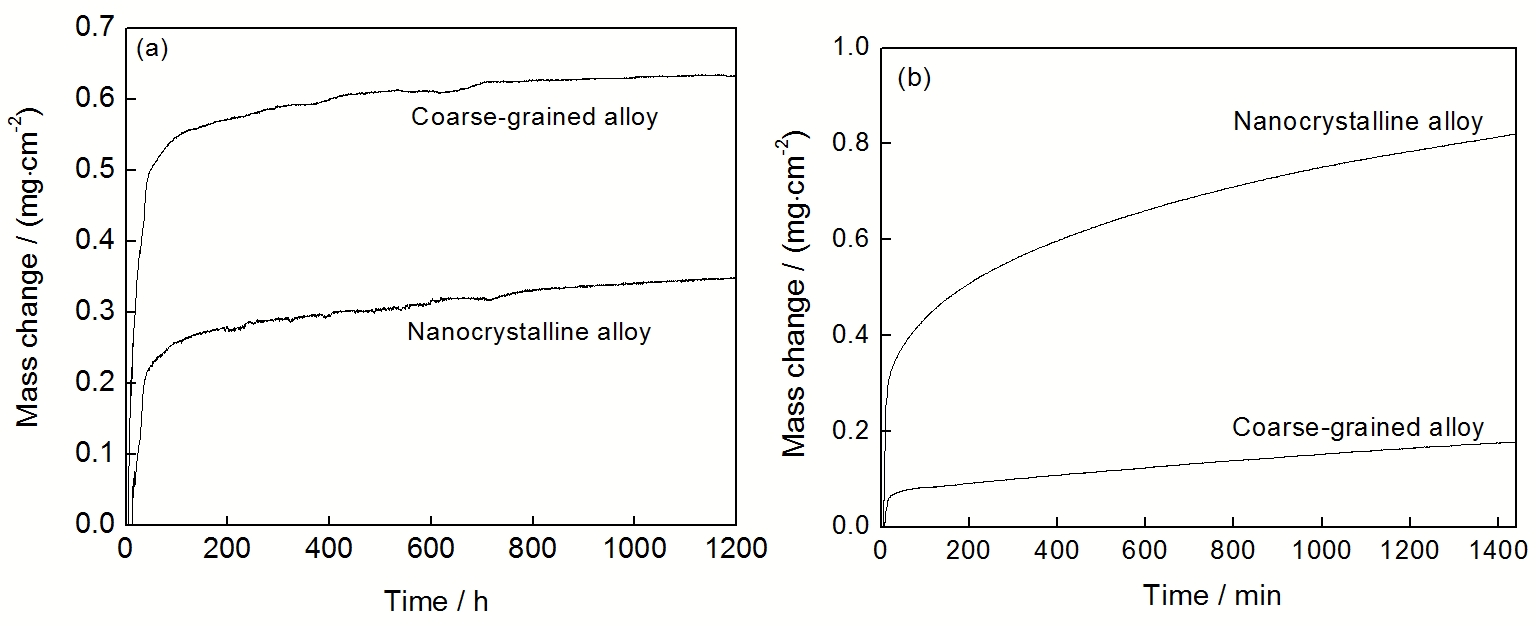
对于能够形成Cr2O3或Al2O3膜的金属材料, 纳米晶化对它们的稳态氧化动力学的影响比较复杂. 例如, Al2O3生长型的粗晶Ni-25Cr-5Al-1S合金经磁控溅射纳米晶化后, 1000 ℃空气中的氧化速率明显降低, 如图5a所示. 这可能与合金纳米晶化促进了Al的选择性氧化, 导致更纯的Al2O3膜(Cr离子等掺杂减少)形成的缘故. 但有些结果刚好相反, 例如, Cr2O3生长型的粗晶304不锈钢经溅射纳米晶化后, 在900 ℃空气中的氧化速率反而升高, 如图5b所示. 该结果可能与以下因素相关, 合金纳米晶化后生长的Cr2O3晶粒度变小, 增多的晶界促进阴、阳离子在膜中的扩散, 从而加速氧化. 因此, 从动力学角度考虑, 保护性氧化膜生长型的金属材料的纳米晶化不一定有利于提高抗氧化性能.
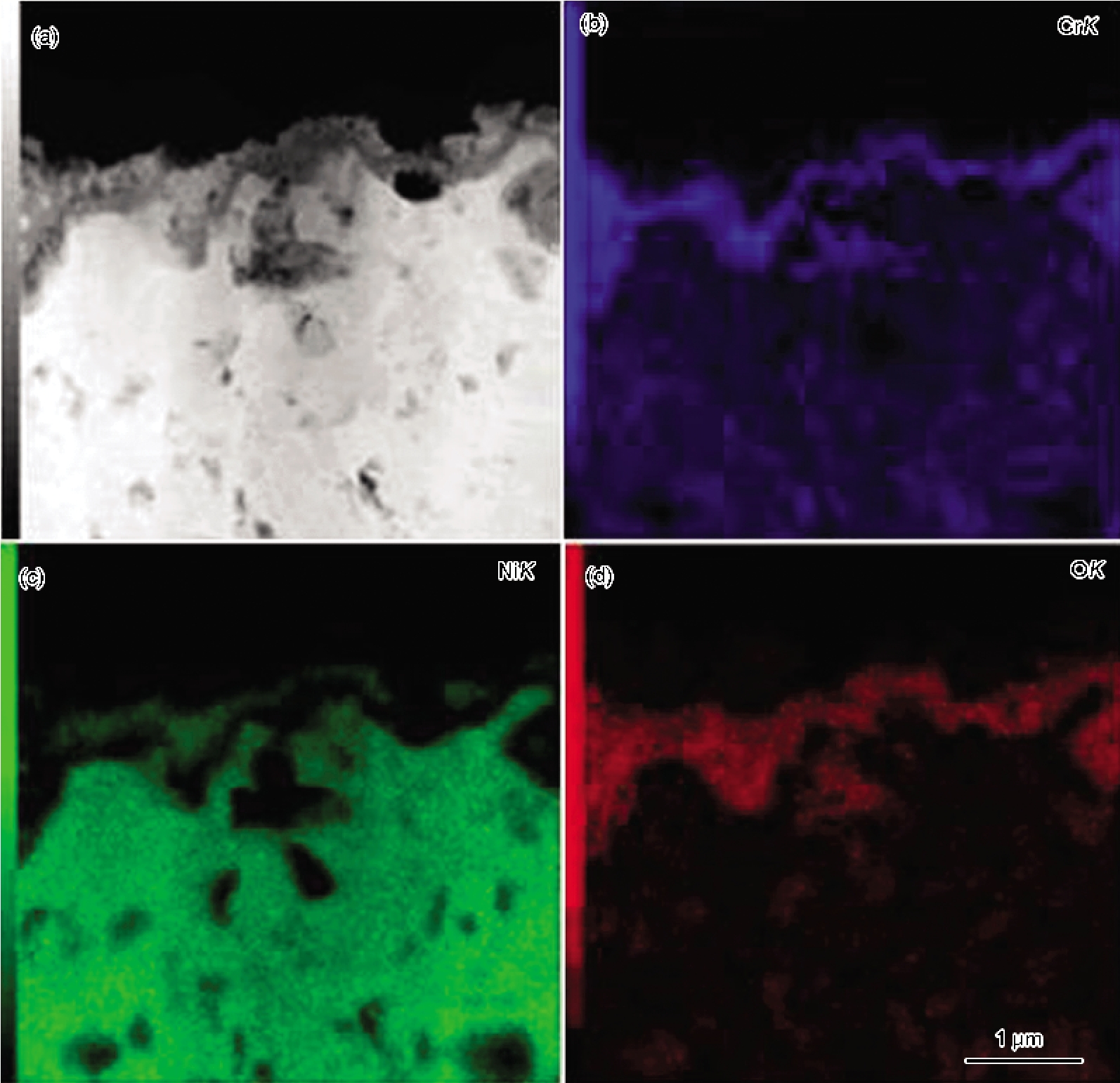
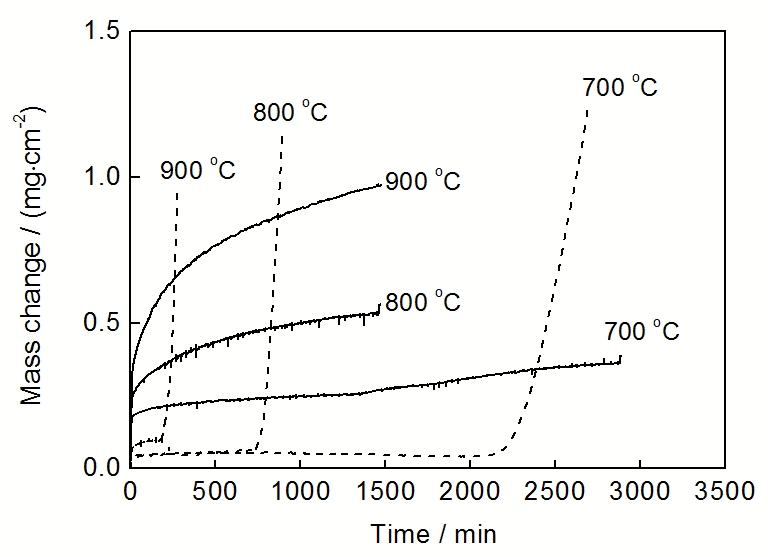
高温氧化时, 粗晶合金生长的保护性氧化膜下易出现成膜元素的贫化区, 当贫化区变宽导致氧化所消耗的量不能立即被补偿时, 保护性氧化膜就不能稳态生长, 即出现失稳氧化, 合金开始生长非保护性氧化物. 失稳氧化易发生于Cr和Al含量低的普通合金以及苛刻氧化环境下. 合金纳米晶化因可避免氧化膜下形成明显的成膜元素的贫化区, 故可防止失稳氧化. 例如, 普通粗晶304不锈钢在干空气中虽能生长Cr2O3膜, 在湿空气中却因Cr2O3的挥发而使得其生长动力学而发生失稳氧化, 但该不锈钢纳米晶化后, 避免了失稳氧化发生[
最后值得一提的是, 由于纳米晶化能通过促进选择性氧化使保护性氧化膜快速形成, 这使得纳米晶金属材料比同化学组成的粗晶合金展现出更佳的抗热腐蚀性能[
抗高温氧化性能的金属材料除了氧化动力学低以外, 热生长氧化膜对金属基体也具有良好的黏附性. 金属材料纳米晶化有利于提高氧化膜的黏附性多有报道, 究其原因, 最常见的解释有如下2种: (1)“钉扎”效应. 氧化物沿晶界向内生长形成许多“微钉”, 把氧化膜固定在金属基体上. 其增强氧化膜黏附性的本质在于通过提高界面粗糙度而提高界面结合能. 因这现象只发现于一些PVD沉积的、在垂直于表面方向为柱状晶的纳米晶金属上, 并且这些氧化物钉子可能因柱状晶之间存在的裂纹所起[
纳米晶金属氧化的力学行为还缺乏系统研究, 金属纳米晶化影响氧化膜黏附性根本原因还有待深入澄清. 实质上, 膜内应力水平、应力释放机制和膜与基体的界面结合强度皆能影响氧化膜黏附性. 多数情况下, 氧化膜的应力水平为热应力和生长应力之和, 但热应力通常远大于生长应力(这也是氧化膜开裂和剥落大多出现在氧化后的降温过程), 膜内应力可通过氧化物蠕变、晶界滑移等降低. 但是, 氧化膜黏附性主要还是取决于界面结合强度, 而后者主要受S在界面的偏聚和空位在界面的沉积影响. 因此, 金属纳米晶化提高氧化膜黏附性的主要原因在于降低或避免了界面S的偏聚以及空位的沉积.
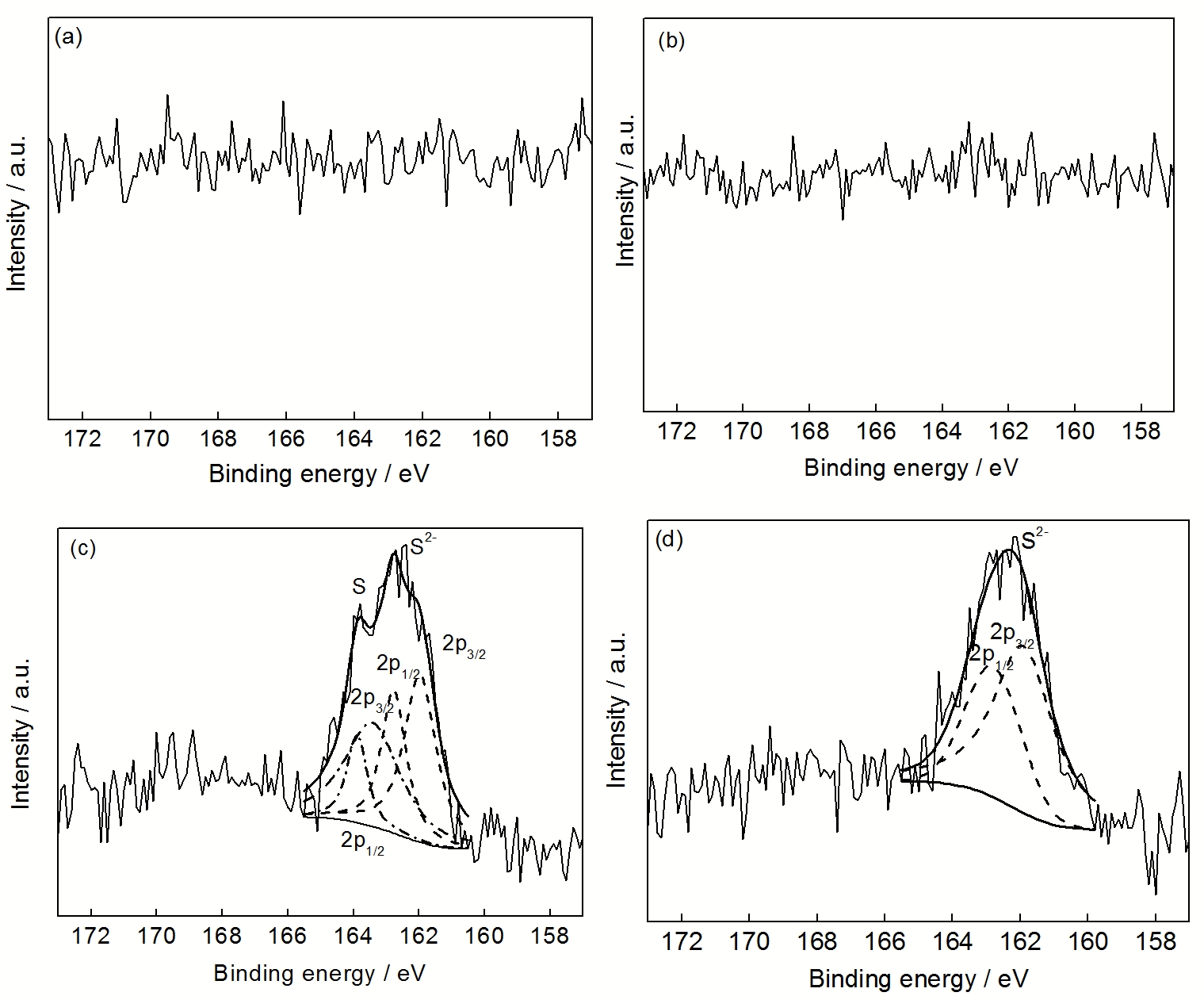
氧化过程中, 金属中的杂质S在界面偏聚, 弱化氧化膜和金属基体界面间的原子键合力, 导致氧化膜黏附性变差, 这一现象被称为“S效应”[
金属纳米晶化后, 丰富的晶界尤其三角晶界可充当S的偏聚场所, 使得S在界面的偏聚浓度降低, 从而增强氧化膜与金属基体的结合力. 这一观点也被高S的粗晶和纳米晶Ni-25Cr-5Al-1S合金的对比循环氧化实验所证实[
氧化过程中, 空位在氧化膜/金属界面沉积形成空洞. 空位源自氧化膜生长过程的阳离子空位, 也可来自Kirkendall空位. 氧化膜下由于成膜元素消耗, 造成其浓度低而非氧化元素浓度高, 从而引起成膜和非成膜元素间的互扩散. 当成膜元素向界面的扩散低于非成膜元素向金属基体内的扩散速度, Kirkendall空位就会在界面沉积. 当界面空洞长大到临界尺寸
式中,
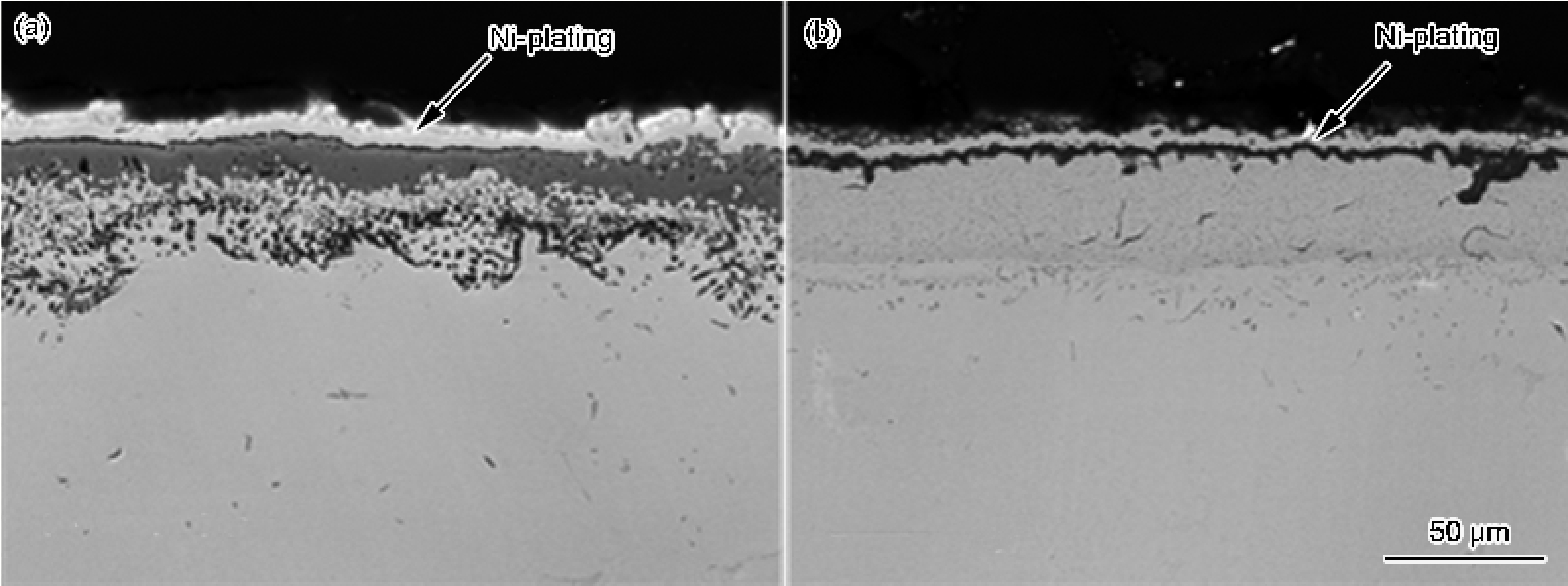
金属晶粒细化和纳米晶化后, 界面及其附近处晶界和三角晶界显著增多, 它们可充当氧化过程中产生的空位湮灭场所, 降低界面空洞的生长动力学, 使氧化膜不易与基体剥离而起皮. 为证实这一观点, 比较了粗晶(晶粒度大于100 mm)和超细晶(晶粒度为325 nm)的Ni3Al抗循环氧化性能[
另外, 由图9b和10可以看到, 粗晶Ni3Al仅热循环5次, 就在Al2O3膜下形成Al的贫化区, 导致界面剥离区再生NiO, 而非更具有保护性的Al2O3膜. 热循环100次后, 贫化区厚度从1 μm增至13 μm[
纳米晶化可使一些Cr和Al含量不高、不具备抗高温腐蚀性的合金热生长保护性Cr2O3或Al2O3膜, 是除施加防护高Cr高Al涂层之外提高金属材料抗高温腐蚀性能的另一防护方法. 此方法可避免高温涂层与金属基材的互扩散而导致涂层的防护性能和基材的力学性能降低. 合金纳米晶化能实现自防护的关键在于促进了Cr或(和)Al的选择性氧化, 利于Cr2O3或Al2O3的形核以及形核后的快速生长. 纳米晶合金中Cr或(和)Al含量、分布均匀性和扩散系数不同, 选择性氧化发生情况及其过程会有所不同. 虽然合金纳米晶在高温下会发生粗化, 但晶粒度很可能维持在比通常的粗晶合金的晶粒度小得多的尺度范围内, 这是Cr2O3或Al2O3生长型的纳米晶合金不易发生失稳氧化以及氧化膜剥离后易再生的原因.
合金纳米晶化利于提高所生长的氧化膜的黏附性, 主要原因可能在于: 合金中尤其氧化膜/合金界面附近处丰富的晶界利于氧化时S的偏聚和空位的沉积, 降低了到达界面的S和空位的浓度或者减少了2者在界面局部区域出现的量值. 排除S的界面毒化效应和避免大量空位在界面沉积的常用方法是在合金中添加少量稀土元素(如Y, Ce, La等)或其氧化物, 纳米晶化在提高金属氧化膜黏附性方面可起与添加稀土类似的效果.
总之, 一些合金的纳米晶化可以实现自防护, 与2个典型的“纳米效应”有关: 即促进保护性氧化膜的形成以及提高氧化膜的黏附性. 纳米晶涂层可呈现与合金纳米晶化相同的纳米效应. 但是, 为纳米晶涂层的应用, 还需要更全面、系统和深入的研究纳米晶的热稳定性对合金的稳态氧化, 保护性氧化膜的再生能力以及氧化膜的力学行为的影响.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|

/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}